Benzonitrile is the chemical compound with the formula C6H5(CN), abbreviated PhCN. This aromatic organic compound is a colorless liquid with a sweet bitter almond odour. It is mainly used as a precursor to the resin benzoguanamine.
The Heck reaction is the chemical reaction of an unsaturated halide with an alkene in the presence of a base and a palladium catalyst to form a substituted alkene. It is named after Tsutomu Mizoroki and Richard F. Heck. Heck was awarded the 2010 Nobel Prize in Chemistry, which he shared with Ei-ichi Negishi and Akira Suzuki, for the discovery and development of this reaction. This reaction was the first example of a carbon-carbon bond-forming reaction that followed a Pd(0)/Pd(II) catalytic cycle, the same catalytic cycle that is seen in other Pd(0)-catalyzed cross-coupling reactions. The Heck reaction is a way to substitute alkenes.
The Suzuki reaction or Suzuki coupling is an organic reaction that uses a palladium complex catalyst to cross-couple a boronic acid to an organohalide. It was first published in 1979 by Akira Suzuki, and he shared the 2010 Nobel Prize in Chemistry with Richard F. Heck and Ei-ichi Negishi for their contribution to the discovery and development of noble metal catalysis in organic synthesis. This reaction is sometimes telescoped with the related Miyaura borylation; the combination is the Suzuki–Miyaura reaction. It is widely used to synthesize polyolefins, styrenes, and substituted biphenyls.
The Simmons–Smith reaction is an organic cheletropic reaction involving an organozinc carbenoid that reacts with an alkene to form a cyclopropane. It is named after Howard Ensign Simmons, Jr. and Ronald D. Smith. It uses a methylene free radical intermediate that is delivered to both carbons of the alkene simultaneously, therefore the configuration of the double bond is preserved in the product and the reaction is stereospecific.
Organopalladium chemistry is a branch of organometallic chemistry that deals with organic palladium compounds and their reactions. Palladium is often used as a catalyst in the reduction of alkenes and alkynes with hydrogen. This process involves the formation of a palladium-carbon covalent bond. Palladium is also prominent in carbon-carbon coupling reactions, as demonstrated in tandem reactions.
The Larock indole synthesis is a heteroannulation reaction that uses palladium as a catalyst to synthesize indoles from an ortho-iodoaniline and a disubstituted alkyne. It is also known as Larock heteroannulation. The reaction is extremely versatile and can be used to produce varying types of indoles. Larock indole synthesis was first proposed by Richard C. Larock in 1991 at Iowa State University.

Organozinc chemistry is the study of the physical properties, synthesis, and reactions of organozinc compounds, which are organometallic compounds that contain carbon (C) to zinc (Zn) chemical bonds.
In organic chemistry, the Buchwald–Hartwig amination is a chemical reaction for the synthesis of carbon–nitrogen bonds via the palladium-catalyzed coupling reactions of amines with aryl halides. Although Pd-catalyzed C–N couplings were reported as early as 1983, Stephen L. Buchwald and John F. Hartwig have been credited, whose publications between 1994 and the late 2000s established the scope of the transformation. The reaction's synthetic utility stems primarily from the shortcomings of typical methods for the synthesis of aromatic C−N bonds, with most methods suffering from limited substrate scope and functional group tolerance. The development of the Buchwald–Hartwig reaction allowed for the facile synthesis of aryl amines, replacing to an extent harsher methods while significantly expanding the repertoire of possible C−N bond formations.

A boronic acid is an organic compound related to boric acid in which one of the three hydroxyl groups is replaced by an alkyl or aryl group. As a compound containing a carbon–boron bond, members of this class thus belong to the larger class of organoboranes.
The Kulinkovich reaction describes the organic synthesis of substituted cyclopropanols through reaction of esters with dialkyldialkoxytitanium reagents, which are generated in situ from Grignard reagents containing a hydrogen in beta-position and titanium(IV) alkoxides such as titanium isopropoxide. This reaction was first reported by Oleg Kulinkovich and coworkers in 1989.

In organometallic chemistry, a metallacycle is a derivative of a carbocyclic compound wherein a metal has replaced at least one carbon center; this is to some extent similar to heterocycles. Metallacycles appear frequently as reactive intermediates in catalysis, e.g. olefin metathesis and alkyne trimerization. In organic synthesis, directed ortho metalation is widely used for the functionalization of arene rings via C-H activation. One main effect that metallic atom substitution on a cyclic carbon compound is distorting the geometry due to the large size of typical metals.
Selenoxide elimination is a method for the chemical synthesis of alkenes from selenoxides. It is most commonly used to synthesize α,β-unsaturated carbonyl compounds from the corresponding saturated analogues. It is mechanistically related to the Cope reaction.
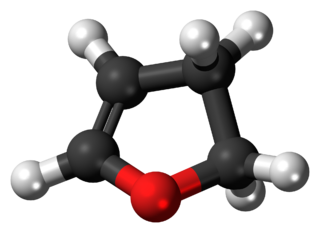
2,3-Dihydrofuran is a heterocyclic compound with the formula C4H6O. It is isomeric with 2,5-dihydrofuran. 2,3-Dihydrofuran is one of the simplest enol ethers. It is a colorless volatile liquid.
Nitrile anions is jargon from the organic product resulting from the deprotonation of alkylnitriles. The proton(s) α to the nitrile group are sufficiently acidic that they undergo deprotonation by strong bases, usually lithium-derived. The products are not anions but covalent organolithium complexes. Regardless, these organolithium compounds are reactive toward various electrophiles.
Trimethylenemethane cycloaddition is the formal (3+2) annulation of trimethylenemethane (TMM) derivatives to two-atom pi systems. Although TMM itself is too reactive and unstable to be stored, reagents which can generate TMM or TMM synthons in situ can be used to effect cycloaddition reactions with appropriate electron acceptors. Generally, electron-deficient pi bonds undergo cyclization with TMMs more easily than electron-rich pi bonds.
Desulfonylation reactions are chemical reactions leading to the removal of a sulfonyl group from organic compounds. As the sulfonyl functional group is electron-withdrawing, methods for cleaving the sulfur–carbon bonds of sulfones are typically reductive in nature. Olefination or replacement with hydrogen may be accomplished using reductive desulfonylation methods.
Metal-catalyzed cyclopropanations are chemical reactions that result in the formation of a cyclopropane ring from a metal carbenoid species and an alkene. In the Simmons–Smith reaction the metal involved is zinc. Metal carbenoid species can be generated through the reaction of a diazo compound with a transition metal). The intramolecular variant of this reaction was first reported in 1961. Rhodium carboxylate complexes, such as dirhodium tetraacetate, are common catalysts. Enantioselective cyclopropanations have been developed.
The Buchner ring expansion is a two-step organic C-C bond forming reaction used to access 7-membered rings. The first step involves formation of a carbene from ethyl diazoacetate, which cyclopropanates an aromatic ring. The ring expansion occurs in the second step, with an electrocyclic reaction opening the cyclopropane ring to form the 7-membered ring.
In organometallic chemistry, the activation of cyclopropanes by transition metals is a research theme with implications for organic synthesis and homogeneous catalysis. Being highly strained, cyclopropanes are prone to oxidative addition to transition metal complexes. The resulting metallacycles are susceptible to a variety of reactions. These reactions are rare examples of C-C bond activation. The rarity of C-C activation processes has been attributed to Steric effects that protect C-C bonds. Furthermore, the directionality of C-C bonds as compared to C-H bonds makes orbital interaction with transition metals less favorable. Thermodynamically, C-C bond activation is more favored than C-H bond activation as the strength of a typical C-C bond is around 90 kcal per mole while the strength of a typical unactivated C-H bond is around 104 kcal per mole.
Jiro Tsuji was a Japanese chemist, notable for his discovery of organometallic reactions, including the Tsuji-Trost reaction, the Tsuji-Wilkinson decarbonylation, and the Tsuji-Wacker reaction.

