
A genetic disorder is a genetic problem caused by one or more abnormalities formed in the genome. Most genetic disorders are quite rare and affect one person in every several thousands or millions. The earliest known genetic condition in a hominid was in the fossil species Paranthropus robustus, with over a third of individuals displaying Amelogenesis imperfecta.

Joubert syndrome is a rare autosomal recessive genetic disorder that affects the cerebellum, an area of the brain that controls balance and coordination.
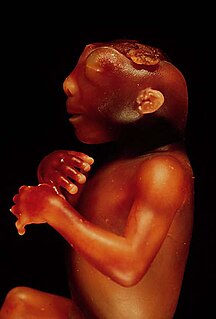
Anencephaly is the absence of a major portion of the brain, skull, and scalp that occurs during embryonic development. It is a cephalic disorder that results from a neural tube defect that occurs when the rostral (head) end of the neural tube fails to close, usually between the 23rd and 26th day following conception. Strictly speaking, the Greek term translates as "no in-head", but it is accepted that children born with this disorder usually only lack a telencephalon, the largest part of the brain consisting mainly of the cerebral hemispheres, including the neocortex, which is responsible for cognition. The remaining structure is usually covered only by a thin layer of membrane—skin, bone, meninges, etc. are all lacking. With very few exceptions, infants with this disorder do not survive longer than a few hours or possibly days after their birth.
Exencephaly, is a type of cephalic disorder wherein the brain is located outside of the skull. This condition is usually found in embryos as an early stage of anencephaly. As an exencephalic pregnancy progresses, the neural tissue gradually degenerates.
Primary ciliary dyskinesia (PCD), is a rare, ciliopathic, autosomal recessive genetic disorder that causes defects in the action of cilia lining the respiratory tract, fallopian tube, and flagellum of sperm cells. The phrase "immotile ciliary syndrome" is no longer favored as the cilia do have movement, but are merely inefficient or unsynchronized.

Ellis–van Creveld syndrome is a rare genetic disorder of the skeletal dysplasia type.
Bardet–Biedl syndrome (BBS) is a ciliopathic human genetic disorder that produces many effects and affects many body systems. It is characterized principally by obesity, retinitis pigmentosa, polydactyly, hypogonadism, and kidney failure in some cases. Historically, slower mental processing has also been considered a principal symptom but is now not regarded as such.
Agenesis of the corpus callosum (ACC) is a rare birth defect in which there is a complete or partial absence of the corpus callosum. It occurs when the corpus callosum, the band of white matter connecting the two hemispheres in the brain, fails to develop normally, typically during pregnancy. The fibers that would otherwise form the corpus callosum become longitudinally oriented along the ipsilateral ventricular wall and form structures called Probst bundles.

Dandy–Walker syndrome (DWS) is a rare group of congenital human brain malformations. There are three subtypes which affect multiple organs to varying degrees, but the fundamental abnormalities involve the cerebellum which controls muscle coordination. The adjacent third and fourth ventricles are often affected, which can alter the flow of cerebrospinal fluid, increase intracranial pressure and lead to multiple other brain function problems.

Alström syndrome (AS), also called Alström–Hallgren syndrome, is a very rare autosomal recessive genetic disorder characterised by childhood obesity and multiple organ dysfunction. Symptoms include early-onset type 2 diabetes, cone-rod dystrophy resulting in blindness, sensorineural hearing loss and dilated cardiomyopathy. Endocrine disorders typically also occur, such as hypergonadotrophic hypogonadism and hypothyroidism, as well as acanthosis nigricans resulting from hyperinsulinemia. Developmental delay is seen in almost half of people with Alström syndrome.

Nephronophthisis is a genetic disorder of the kidneys which affects children. It is classified as a medullary cystic kidney disease. The disorder is inherited in an autosomal recessive fashion and, although rare, is the most common genetic cause of childhood kidney failure. It is a form of ciliopathy. Its incidence has been estimated to be 0.9 cases per million people in the United States, and 1 in 50,000 births in Canada.
Senior–Løken syndrome is a congenital eye disorder, first characterized in 1961. It is a rare, ciliopathic, autosomal recessive disorder characterized by juvenile nephronophthis and progressive eye disease.

A ciliopathy is a genetic disorder of the cellular cilia or the cilia anchoring structures, the basal bodies, or of ciliary function.
Marden–Walker syndrome (MWS) is a rare autosomal recessive congenital disorder. It is characterized by blepharophimosis, microcephaly, micrognathia, multiple joint contractures, arachnodactyly, camptodactyly, kyphoscoliosis, and delayed motor development and is often associated with cystic dysplastic kidneys, dextrocardia, Dandy-Walker malformation, and agenesis of corpus callosum".
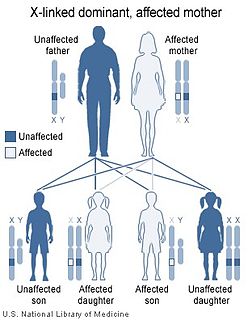
Orofaciodigital syndrome 1 (OFD1), also called Papillon-League and Psaume syndrome, is an X-linked congenital disorder characterized by malformations of the face, oral cavity, and digits with polycystic kidney disease and variable involvement of the central nervous system.
Hydrolethalus syndrome (HLS) is a rare genetic disorder that causes improper fetal development, resulting in birth defects and, most commonly, stillbirth.

Meckel syndrome, type 1 also known as MKS1 is a protein that in humans is encoded by the MKS1 gene.
A Finnish heritage disease is a genetic disease or disorder that is significantly more common in people whose ancestors were ethnic Finns, natives of Finland and Sweden (Meänmaa) and Russia. 36 rare diseases are regarded as Finnish heritage diseases. The diseases are not restricted to Finns; they are genetic diseases with far wider distribution in the world, but due to founder effects and genetic isolation they are more common in Finns.
McKusick–Kaufman syndrome is a genetic condition associated with MKKS.
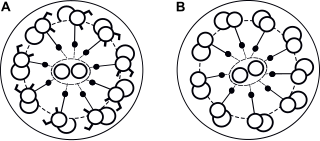
Ciliogenesis is defined as the building of the cell's antenna or extracellular fluid mediation mechanism. It includes the assembly and disassembly of the cilia during the cell cycle. Cilia are important organelles of cells and are involved in numerous activities such as cell signaling, processing developmental signals, and directing the flow of fluids such as mucus over and around cells. Due to the importance of these cell processes, defects in ciliogenesis can lead to numerous human diseases related to non-functioning cilia. Ciliogenesis may also play a role in the development of left/right handedness in humans.
