An ylide or ylid is a neutral dipolar molecule containing a formally negatively charged atom (usually a carbanion) directly attached to a heteroatom with a formal positive charge (usually nitrogen, phosphorus or sulfur), and in which both atoms have full octets of electrons. The result can be viewed as a structure in which two adjacent atoms are connected by both a covalent and an ionic bond; normally written X+–Y−. Ylides are thus 1,2-dipolar compounds, and a subclass of zwitterions. They appear in organic chemistry as reagents or reactive intermediates.

Tetrahedrane is a hypothetical platonic hydrocarbon with chemical formula C4H4 and a tetrahedral structure. The molecule would be subject to considerable angle strain and has not been synthesized as of 2023. However, a number of derivatives have been prepared. In a more general sense, the term tetrahedranes is used to describe a class of molecules and ions with related structure, e.g. white phosphorus.
In chemistry, a hypervalent molecule is a molecule that contains one or more main group elements apparently bearing more than eight electrons in their valence shells. Phosphorus pentachloride, sulfur hexafluoride, chlorine trifluoride, the chlorite ion, and the triiodide ion are examples of hypervalent molecules.

In chemistry, a phosphaalkyne is an organophosphorus compound containing a triple bond between phosphorus and carbon with the general formula R-C≡P. Phosphaalkynes are the heavier congeners of nitriles, though, due to the similar electronegativities of phosphorus and carbon, possess reactivity patterns reminiscent of alkynes. Due to their high reactivity, phosphaalkynes are not found naturally on earth, but the simplest phosphaalkyne, phosphaethyne (H-C≡P) has been observed in the interstellar medium.
Diphosphorus is an inorganic chemical with the chemical formula P
2. Unlike nitrogen, its lighter pnictogen neighbor which forms a stable N2 molecule with a nitrogen to nitrogen triple bond, phosphorus prefers a tetrahedral form P4 because P-P pi-bonds are high in energy. Diphosphorus is, therefore, very reactive with a bond-dissociation energy (117 kcal/mol or 490 kJ/mol) half that of dinitrogen. The bond distance has been measured at 1.8934 Å.

Phosphinidenes are low-valent phosphorus compounds analogous to carbenes and nitrenes, having the general structure RP. The "free" form of these compounds is conventionally described as having a singly-coordinated phosphorus atom containing only 6 electrons in its valence level. Most phosphinidenes are highly reactive and short-lived, thereby complicating empirical studies on their chemical properties. In the last few decades, several strategies have been employed to stabilize phosphinidenes, and researchers have developed a number of reagents and systems that can generate and transfer phosphinidenes as reactive intermediates in the synthesis of various organophosphorus compounds.

tert-Butylphosphaacetylene is an organophosphorus compound. Abbreviated t-BuCP, it was the first example of an isolable phosphaalkyne. Prior to its synthesis, the double bond rule had suggested that elements of Period 3 and higher were unable to form double or triple bonds with lighter main group elements because of weak orbital overlap. The synthesis of t-BuCP discredited much of the double bond rule and opened new studies into the formation of unsaturated phosphorus compounds.

Tetranitrogen is a neutrally charged polynitrogen allotrope of the chemical formula N
4 and consists of four nitrogen atoms. The tetranitrogen cation is the positively charged ion, N+
4, which is more stable than the neutral tetranitrogen molecule and is thus more studied.

Phosphorus mononitride is an inorganic compound with the chemical formula PN. Containing only phosphorus and nitrogen, this material is classified as a binary nitride. From the Lewis structure perspective, it can be represented with a P-N triple bond with a lone pair on each atom. It is isoelectronic with N2, CO, P2, CS and SiO.
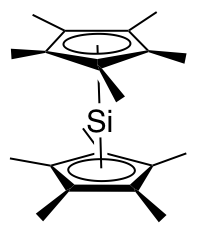
Decamethylsilicocene, (C5Me5)2Si, is a group 14 sandwich compound. It is an example of a main-group cyclopentadienyl complex; these molecules are related to metallocenes but contain p-block elements as the central atom. It is a colorless, air sensitive solid that sublimes under vacuum.
A phosphetane is a 4-membered organophosphorus heterocycle. The parent phosphetane molecule, which has the formula C3H7P, is one atom larger than phosphiranes, one smaller than phospholes, and is the heavy-atom analogue of azetidines. The first known phosphetane synthesis was reported in 1957 by Kosolapoff and Struck, but the method was both inefficient and hard to reproduce, with yields rarely exceeding 1%. A far more efficient method was reported in 1962 by McBride, whose method allowed for the first studies into the physical and chemical properties of phosphetanes. Phosphetanes are a well understood class of molecules that have found broad applications as chemical building blocks, reagents for organic/inorganic synthesis, and ligands in coordination chemistry.

Phosphasilenes or silylidenephosphanes are a class of compounds with silicon-phosphorus double bonds. Since the electronegativity of phosphorus (2.1) is higher than that of silicon (1.9), the "Si=P" moiety of phosphasilene is polarized. The degree of polarization can be tuned by altering the coordination numbers of the Si and P centers, or by modifying the electronic properties of the substituents. The phosphasilene Si=P double bond is highly reactive, yet with the choice of proper substituents, it can be stabilized via donor-acceptor interaction or by steric congestion.

An N-Heterocyclic silylene (NHSi) is an uncharged heterocyclic chemical compound consisting of a divalent silicon atom bonded to two nitrogen atoms. The isolation of the first stable NHSi, also the first stable dicoordinate silicon compound, was reported in 1994 by Michael Denk and Robert West three years after Anthony Arduengo first isolated an N-heterocyclic carbene, the lighter congener of NHSis. Since their first isolation, NHSis have been synthesized and studied with both saturated and unsaturated central rings ranging in size from 4 to 6 atoms. The stability of NHSis, especially 6π aromatic unsaturated five-membered examples, make them useful systems to study the structure and reactivity of silylenes and low-valent main group elements in general. Though not used outside of academic settings, complexes containing NHSis are known to be competent catalysts for industrially important reactions. This article focuses on the properties and reactivity of five-membered NHSis.

Nontrigonal pnictogen compounds refer to tricoordinate trivalent pnictogen compounds that are not of typical trigonal pyramidal molecular geometry. By virtue of their geometric constraint, these compounds exhibit distinct electronic structures and reactivities, which bestow on them potential to provide unique nonmetal platforms for bond cleavage reactions.

Arsenic in the solid state can be found as gray, black, or yellow allotropes. These various forms feature diverse structural motifs, with yellow arsenic enabling the widest range of reactivity. In particular, reaction of yellow arsenic with main group and transition metal elements results in compounds with wide-ranging structural motifs, with butterfly, sandwich and realgar-type moieties featuring most prominently.
1-Phosphaallenes is are allenes in which the first carbon atom is replaced by phosphorus, resulting in the structure: -P=C=C<.
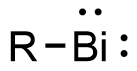
Bismuthinidenes are a class of organobismuth compounds, analogous to carbenes. These compounds have the general form R-Bi, with two lone pairs of electrons on the central bismuth(I) atom. Due to the unusually low valency and oxidation state of +1, most bismuthinidenes are reactive and unstable, though in recent decades, both transition metals and polydentate chelating Lewis base ligands have been employed to stabilize the low-valent bismuth(I) center through steric protection and π donation either in solution or in crystal structures. Lewis base-stabilized bismuthinidenes adopt a singlet ground state with an inert lone pair of electrons in the 6s orbital. A second lone pair in a 6p orbital and a single empty 6p orbital make Lewis base-stabilized bismuthinidenes ambiphilic.
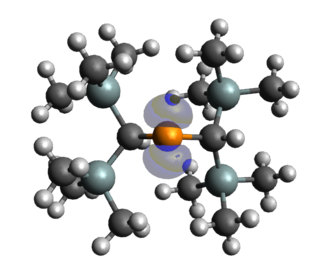
Stable and persistent phosphorus radicals are phosphorus-centred radicals that are isolable and can exist for at least short periods of time. Radicals consisting of main group elements are often very reactive and undergo uncontrollable reactions, notably dimerization and polymerization. The common strategies for stabilising these phosphorus radicals usually include the delocalisation of the unpaired electron over a pi system or nearby electronegative atoms, and kinetic stabilisation with bulky ligands. Stable and persistent phosphorus radicals can be classified into three categories: neutral, cationic, and anionic radicals. Each of these classes involve various sub-classes, with neutral phosphorus radicals being the most extensively studied. Phosphorus exists as one isotope 31P (I = 1/2) with large hyperfine couplings relative to other spin active nuclei, making phosphorus radicals particularly attractive for spin-labelling experiments.
Phosphiranes are organic compounds with the phosphirane functional group – a three-membered ring with two atoms of carbon and one atom of phosphorus that has lots of ring angle strain. Phosphiranes are usually synthesized by double substitution reactions or pericyclic pathways. Phosphiranes can also be oxidized into phosphirane oxides, undergo SN2 substitution reactions, or decompose into different units.

Tris(silox)tantalum, Ta(SiOtBu)3, is an organotantalum complex bound with three siloxide (silox="tBu3SiO-") ligands,. The tantalum center has a d-electron count of 2 and an oxidation state of III. The complex is trigonal planar whose point group is assigned as D3h. It is a crystalline light blue solid which forms blue-green solutions in THF.















. Diphosphatetrahedrane Ligand Substitution with Silver Complex.png](http://upload.wikimedia.org/wikipedia/commons/thumb/a/ad/Diphosphatetrahedrane_Ligand_Substitution_with_Silver_Complex.png/500px-Diphosphatetrahedrane_Ligand_Substitution_with_Silver_Complex.png)







