Related Research Articles

Kabuki syndrome is a rare congenital disorder of genetic origin. It affects multiple parts of the body, with varying symptoms and severity, although the most common is the characteristic facial appearance.

ICF syndrome is a very rare autosomal recessive immune disorder.
Cohen syndrome is a very rare autosomal recessive genetic disorder with varied expression, characterised by obesity, intellectual disability, distinct craniofacial abnormalities and potential ocular dysfunction.
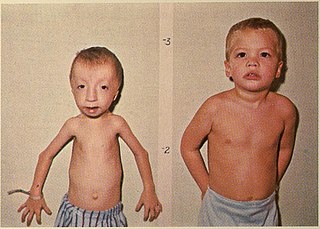
Seckel syndrome, or microcephalic primordial dwarfism is an extremely rare congenital nanosomic disorder. Inheritance is autosomal recessive. It is characterized by intrauterine growth restriction and postnatal dwarfism with a small head, narrow bird-like face with a beak-like nose, large eyes with down-slanting palpebral fissures, receding mandible and intellectual disability.

Duane-radial ray syndrome, also known as Okihiro syndrome, is a rare autosomal dominant disorder that primarily affects the eyes and causes abnormalities of bones in the arms and hands. This disorder is considered to be a SALL4-related disorder due to the SALL4 gene mutations leading to these abnormalities. It is diagnosed by clinical findings on a physical exam as well as genetic testing and imaging. After being diagnosed, there are other evaluations that one may go through in order to determine the extent of the disease. There are various treatments for the symptoms of this disorder.

Zimmermann–Laband syndrome (ZLS) is two different conditions that share similar clinical features. It is an extremely rare, autosomal dominant congenital disorder.
Young–Simpson syndrome (YSS) is a rare congenital disorder with symptoms including hypothyroidism, heart defects, facial dysmorphism, cryptorchidism in males, hypotonia, intellectual disability, and postnatal growth retardation.
Weaver syndrome is an extremely rare autosomal dominant genetic disorder associated with rapid growth beginning in the prenatal period and continuing through the toddler and youth years. It is characterized by advanced osseous maturation and distinctive craniofacial, skeletal and neurological abnormalities. It is similar to Sotos syndrome and is classified as an overgrowth syndrome.

DNA (cytosine-5)-methyltransferase 3A (DNMT3A) is an enzyme that catalyzes the transfer of methyl groups to specific CpG structures in DNA, a process called DNA methylation. The enzyme is encoded in humans by the DNMT3A gene.

Probable E3 ubiquitin-protein ligase HERC1 is an enzyme that in humans is encoded by the HERC1 gene.
Trichothiodystrophy (TTD) is an autosomal recessive inherited disorder characterised by brittle hair and intellectual impairment. The word breaks down into tricho – "hair", thio – "sulphur", and dystrophy – "wasting away" or literally "bad nourishment". TTD is associated with a range of symptoms connected with organs of the ectoderm and neuroectoderm. TTD may be subclassified into four syndromes: Approximately half of all patients with trichothiodystrophy have photosensitivity, which divides the classification into syndromes with or without photosensitivity; BIDS and PBIDS, and IBIDS and PIBIDS. Modern covering usage is TTD-P (photosensitive), and TTD.

Sotos syndrome is a rare genetic disorder characterized by excessive physical growth during the first years of life. Excessive growth often starts in infancy and continues into the early teen years. The disorder may be accompanied by autism, mild intellectual disability, delayed motor, cognitive, and social development, hypotonia, and speech impairments. Children with Sotos syndrome tend to be large at birth and are often taller, heavier, and have relatively large skulls (macrocephaly) than is normal for their age. Signs of the disorder, which vary among individuals, include a disproportionately large skull with a slightly protrusive forehead, large hands and feet, large mandible, hypertelorism, and downslanting eyes. Clumsiness, an awkward gait, and unusual aggressiveness or irritability may also occur.
Progeroid syndromes (PS) are a group of rare genetic disorders that mimic physiological aging, making affected individuals appear to be older than they are. The term progeroid syndrome does not necessarily imply progeria, which is a specific type of progeroid syndrome.
Wiedemann–Rautenstrauch (WR) syndrome, also known as neonatal progeroid syndrome, is a rare autosomal recessive progeroid syndrome. There have been over 30 cases of WR. WR is associated with abnormalities in bone maturation, and lipids and hormone metabolism.

Jean-Louis Mandel, born in Strasbourg on February 12, 1946, is a French medical doctor and geneticist, and heads a research team at the Institute of Genetics and Molecular and Cellular Biology (IGBMC). He has been in charge of the genetic diagnosis laboratory at the University Hospitals of Strasbourg since 1992, as well as a professor at the Collège de France since 2003.
SYNGAP1-related intellectual disability is a monogenetic developmental and epileptic encephalopathy that affects the central nervous system. Symptoms include intellectual disability, epilepsy, autism, sensory processing deficits, hypotonia and unstable gait.

Temple–Baraitser syndrome (TBS) is a very rare autosomal dominant genetic disorder, characterised by intellectual disability, epilepsy, small or absent nail of the thumbs and great toes, and distinct craniofacial features.
Autosomal dominant intellectual disability-craniofacial anomalies-cardiac defects syndrome is a rare genetic disorder which is characterized by multi-systemic symptoms primarily affecting the intellect and post-natal development.
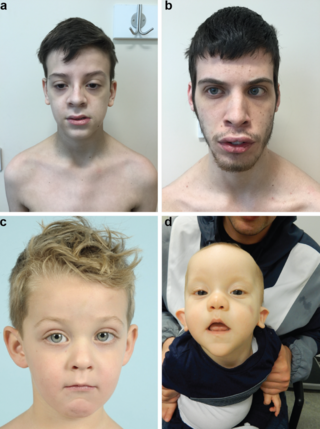
Beck–Fahrner syndrome, also known as BEFAHRS and TET3 deficiency, is a rare genetic disorder caused by mutations of the TET3 gene. The clinical presentation varies among individuals, but typically includes global developmental delay, slow progress in mental and physical activities, autism, decreased muscle tone, epilepsy and dysmorphic features.
Malan syndrome (MALNS) is a rare overgrowth syndrome caused by autosomal dominant mutations in the NFIX gene. The syndrome is characterized by overgrowth, craniofacial dysmorphia, intellectual disability, and behavioral issues. It was formerly diagnosed as Sotos syndrome 2.
References
- 1 2 3 4 5 6 Ostrowski PJ, Tatton-Brown K (2022-06-30). Adam MP, Feldman J, Mirzaa GM, et al. (eds.). "Tatton-Brown-Rahman Syndrome". GeneReviews. University of Washington, Seattle. PMID 35771960 . Retrieved 2024-01-09.
- 1 2 3 Tatton-Brown K, Seal S, Ruark E, Harmer J, Ramsay E, et al. (2014). "Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability". Nat Genet. 46 (4): 385–8. doi:10.1038/ng.2917. PMC 3981653 . PMID 24614070.
- 1 2 Tatton-Brown K, Zachariou A, Loveday C, Renwick A, Mahamdallie S, et al. (2018). "The Tatton-Brown-Rahman Syndrome: A clinical study of 55 individuals with de novo constitutive DNMT3A variants". Wellcome Open Research. 3: 46. doi: 10.12688/wellcomeopenres.14430.1 . PMC 5964628 . PMID 29900417.
- ↑ Jiménez de la Peña M, Rincón-Pérez I, López-Martín S, Albert J, Martín Fernández-Mayoralas D, Fernández-Perrone AL, et al. (2023). "Tatton-Brown-Rahman syndrome: Novel pathogenic variants and new neuroimaging findings". Am J Med Genet A. 194 (2): 211–217. doi: 10.1002/ajmg.a.63434 . PMID 37795572.
- ↑ Hamagami N, Wu DY, Clemens AW, Nettles SA, Li A, Gabel HW (2023). "NSD1 deposits histone H3 lysine 36 dimethylation to pattern non-CG DNA methylation in neurons". Mol Cell. 83 (9): 1412–1428.e7. doi:10.1016/j.molcel.2023.04.001. PMC 10230755 . PMID 37098340.