
Alport syndrome is a genetic disorder affecting around 1 in 5,000-10,000 children, characterized by glomerulonephritis, end-stage kidney disease, and hearing loss. Alport syndrome can also affect the eyes, though the changes do not usually affect vision, except when changes to the lens occur in later life. Blood in urine is universal. Proteinuria is a feature as kidney disease progresses.

Hurler syndrome, also known as mucopolysaccharidosis Type IH (MPS-IH), Hurler's disease, and formerly gargoylism, is a genetic disorder that results in the buildup of large sugar molecules called glycosaminoglycans (GAGs) in lysosomes. The inability to break down these molecules results in a wide variety of symptoms caused by damage to several different organ systems, including but not limited to the nervous system, skeletal system, eyes, and heart.

Coffin–Lowry syndrome is a genetic disorder that is X-linked dominant and which causes severe mental problems sometimes associated with abnormalities of growth, cardiac abnormalities, kyphoscoliosis, as well as auditory and visual abnormalities.

Rubinstein–Taybi syndrome (RTS) is a rare genetic condition characterized by short stature, moderate to severe learning difficulties, distinctive facial features, and broad thumbs and first toes. Other features of the disorder vary among affected individuals. These characteristics are caused by a mutation or deletion in the CREBBP gene, located on chromosome 16, and/or the EP300 gene, located on chromosome 22.
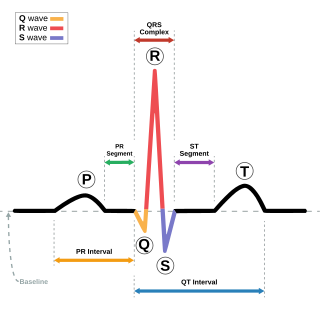
Andersen–Tawil syndrome, also called Andersen syndrome and long QT syndrome 7, is a rare genetic disorder affecting several parts of the body. The three predominant features of Andersen–Tawil syndrome include disturbances of the electrical function of the heart characterised by an abnormality seen on an electrocardiogram and a tendency to abnormal heart rhythms, physical characteristics including low-set ears and a small lower jaw, and intermittent periods of muscle weakness known as hypokalaemic periodic paralysis.

Cohen syndrome is a very rare autosomal recessive genetic disorder with varied expression, characterised by obesity, intellectual disability, distinct craniofacial abnormalities and potential ocular dysfunction.

Alpha-thalassemia mental retardation syndrome (ATRX), also called alpha-thalassemia X-linked intellectual disability syndrome, nondeletion type or ATR-X syndrome, is an X-linked recessive condition associated with a mutation in the ATRX gene. Males with this condition tend to be moderately intellectually disabled and have physical characteristics including coarse facial features, microcephaly, hypertelorism, a depressed nasal bridge, a tented upper lip and an everted lower lip. Mild or moderate anemia, associated with alpha-thalassemia, is part of the condition. Females with this mutated gene have no specific signs or features, but if they do, they may demonstrate skewed X chromosome inactivation.
Abruzzo–Erickson syndrome is an extremely rare disorder characterized by deafness, protruding ears, coloboma, a cleft palate or palatal rugosity, radial synostosis, and short stature. It was first characterized by Abruzzo and Erickson in 1977 as a CHARGE like syndrome as variably expressed among a family of two brothers, their mother, and their maternal uncle. Members of this family exhibited many of the CHARGE symptoms, but notably did not have choanal atresia and the brothers experienced typical genital development. Due to the recent discovery of this disorder, its etiology is not fully known but it is understood that it arises from mutations on the TBX22 gene on the X-chromosome. The disorder is inherited in an X-linked recessive manner. There is currently no known cure but its symptoms can be treated.

Alström syndrome (AS), also called Alström–Hallgren syndrome, is a very rare autosomal recessive genetic disorder characterised by childhood obesity and multiple organ dysfunction. Symptoms include early-onset type 2 diabetes, cone-rod dystrophy resulting in blindness, sensorineural hearing loss and dilated cardiomyopathy. Endocrine disorders typically also occur, such as hypergonadotrophic hypogonadism and hypothyroidism, as well as acanthosis nigricans resulting from hyperinsulinemia. Developmental delay is seen in almost half of people with Alström syndrome.
Aarskog–Scott syndrome (AAS) is a rare disease inherited as X-linked and characterized by short stature, facial abnormalities, skeletal and genital anomalies. This condition mainly affects males, although females may have mild features of the syndrome.

Young–Simpson syndrome (YSS) is a rare congenital disorder with symptoms including hypothyroidism, heart defects, facial dysmorphism, cryptorchidism in males, hypotonia, intellectual disability, and postnatal growth retardation.
Weaver syndrome is a rare autosomal dominant genetic disorder associated with rapid growth beginning in the prenatal period and continuing through the toddler and youth years. It is characterized by advanced osseous maturation and distinctive craniofacial, skeletal and neurological abnormalities. It is similar to Sotos syndrome and is classified as an overgrowth syndrome.
Paroxysmal extreme pain disorder originally named familial rectal pain syndrome, is a rare disorder whose most notable features are pain in the mandibular, ocular and rectal areas as well as flushing. PEPD often first manifests at the beginning of life, perhaps even in utero, with symptoms persisting throughout life. PEPD symptoms are reminiscent of primary erythromelalgia, as both result in flushing and episodic pain, though pain is typically present in the extremities for primary erythromelalgia. Both of these disorders have recently been shown to be allelic, both caused by mutations in the voltage-gated sodium channel NaV1.7 encoded by the gene SCN9A. A different mutation in the SCN9A ion channel causes congenital insensitivity to pain.

Intermembrane lipid transfer protein VPS13B, also known as vacuolar protein sorting-associated 13B, and Cohen syndrome protein 1 is a protein that in humans is encoded by the VPS13B gene. It is a giant protein associated with the Golgi apparatus that is believed to be involved in post-Golgi apparatus sorting and trafficking. Mutations in the human VPS13B gene cause Cohen syndrome.

Winchester syndrome is a rare hereditary connective tissue disease described in 1969, of which the main characteristics are short stature, marked contractures of joints, opacities in the cornea, coarse facial features, dissolution of the carpal and tarsal bones, and osteoporosis. Winchester syndrome was once considered to be related to a similar condition, multicentric osteolysis, nodulosis, and arthropathy (MONA). However, it was discovered that the two are caused by mutations found in different genes; however they mostly produce the same phenotype or clinical picture. Appearances resemble rheumatoid arthritis. Increased uronic acid is demonstrated in cultured fibroblasts from the skin and to a lesser degree in both parents. Despite initial tests not showing increased mucopolysaccharide excretion, the disease was regarded as a mucopolysaccharidosis. Winchester syndrome is thought to be inherited as an autosomal recessive trait.

Lujan–Fryns syndrome (LFS) is an X-linked genetic disorder that causes mild to moderate intellectual disability and features described as Marfanoid habitus, referring to a group of physical characteristics similar to those found in Marfan syndrome. These features include a tall, thin stature and long, slender limbs. LFS is also associated with psychopathology and behavioral abnormalities, and it exhibits a number of malformations affecting the brain and heart. The disorder is inherited in an X-linked dominant manner, and is attributed to a missense mutation in the MED12 gene. There is currently no treatment or therapy for the underlying MED12 malfunction, and the exact cause of the disorder remains unclear.
Parkes Weber syndrome (PWS) is a congenital disorder of the vascular system. It is an extremely rare condition, and its exact prevalence is unknown. It is named after British dermatologist Frederick Parkes Weber, who first described the syndrome in 1907.

Sotos syndrome is a rare genetic disorder characterized by excessive physical growth during the first years of life. Excessive growth often starts in infancy and continues into the early teen years. The disorder may be accompanied by autism, mild intellectual disability, delayed motor, cognitive, and social development, hypotonia, and speech impairments. Children with Sotos syndrome tend to be large at birth and are often taller, heavier, and have relatively large skulls (macrocephaly) than is normal for their age. Signs of the disorder, which vary among individuals, include a disproportionately large skull with a slightly protrusive forehead, large hands and feet, large mandible, hypertelorism, and downslanting eyes. Clumsiness, an awkward gait, and unusual aggressiveness or irritability may also occur.
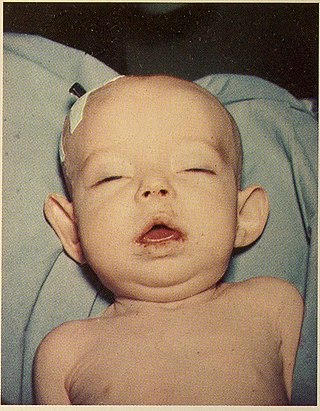
Donohue syndrome is an extremely rare and severe genetic disorder. Leprechaunism derives its name from the hallmark elvish features exhibited by the affected individuals. The disease is caused by a mutation in the INSR gene, which contains the genetic information for the formation of insulin receptors. As a result, affected individuals have either a decreased number of insulin receptors, or insulin receptor with greatly impaired functionality. The lack and impairment of insulin receptor functionality leads to an inability to regulate blood glucose levels through severe insulin resistance. This will ultimately lead to affected development of tissues and organs throughout the body. In addition to the physical abnormalities, leprechaunism is also characterized by endocrine system abnormalities that can lead to conditions such as hyperglycemia, hypoglycemia, hyperinsulemia, and the enlargement of certain sex organs such as the penis in males, and the clitoris in females.
Roussy–Lévy syndrome, also known as Roussy–Lévy areflexic dystasia, is a rare disorder of humans that results in progressive muscle wasting. It is caused by mutation the s that code for proteins necessary for the functioning of the myelin sheath of the, affecting the conductance of nerve signals and resulting in loss of muscles' ability to move.
