
Angiotensin is a peptide hormone that causes vasoconstriction and an increase in blood pressure. It is part of the renin–angiotensin system, which regulates blood pressure. Angiotensin also stimulates the release of aldosterone from the adrenal cortex to promote sodium retention by the kidneys.
The angiotensin II receptors, (ATR1) and (ATR2), are a class of G protein-coupled receptors with angiotensin II as their ligands. They are important in the renin–angiotensin system: they are responsible for the signal transduction of the vasoconstricting stimulus of the main effector hormone, angiotensin II.

Hepatocyte growth factor receptor is a protein that in humans is encoded by the MET gene. The protein possesses tyrosine kinase activity. The primary single chain precursor protein is post-translationally cleaved to produce the alpha and beta subunits, which are disulfide linked to form the mature receptor.

Norbuprenorphine is a major active metabolite of the opioid modulator buprenorphine. It is a μ-opioid, δ-opioid, and nociceptin receptor full agonist, and a κ-opioid receptor partial agonist. In rats, unlike buprenorphine, norbuprenorphine produces marked respiratory depression but with very little antinociceptive effect. In explanation of these properties, norbuprenorphine has been found to be a high affinity P-glycoprotein substrate, and in accordance, shows very limited blood-brain-barrier penetration.

Hepatocyte growth factor (HGF) or scatter factor (SF) is a paracrine cellular growth, motility and morphogenic factor. It is secreted by mesenchymal cells and targets and acts primarily upon epithelial cells and endothelial cells, but also acts on haemopoietic progenitor cells and T cells. It has been shown to have a major role in embryonic organ development, specifically in myogenesis, in adult organ regeneration, and in wound healing.
Neuromedin U is a neuropeptide found in the brain of humans and other mammals, which has a number of diverse functions including contraction of smooth muscle, regulation of blood pressure, pain perception, appetite, bone growth, and hormone release. It was first isolated from the spinal cord in 1985, and named after its ability to cause smooth muscle contraction in the uterus.

HNF1 homeobox A, also known as HNF1A, is a human gene on chromosome 12. It is ubiquitously expressed in many tissues and cell types. The protein encoded by this gene is a transcription factor that is highly expressed in the liver and is involved in the regulation of the expression of several liver-specific genes. Mutations in the HNF1A gene have been known to cause diabetes. The HNF1A gene also contains a SNP associated with increased risk of coronary artery disease.
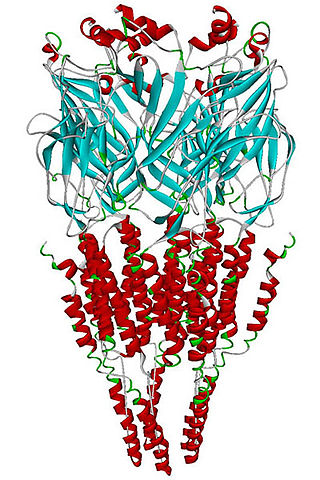
The alpha-7 nicotinic receptor, also known as the α7 receptor, is a type of nicotinic acetylcholine receptor implicated in long-term memory, consisting entirely of α7 subunits. As with other nicotinic acetylcholine receptors, functional α7 receptors are pentameric [i.e., (α7)5 stoichiometry].

LY-503430 is an AMPA receptor positive allosteric modulator developed by Eli Lilly.

Neramexane is a drug related to memantine, which acts as an NMDA antagonist and has neuroprotective effects. It is being developed for various possible applications, including treatment of tinnitus, Alzheimer's disease, drug addiction and as an analgesic. Animal studies have also suggested antidepressant and nootropic actions, so there are a wide range of potential applications this drug may be used for. It also acts as a nicotinic acetylcholine receptor antagonist.

ICI-118,551 is a selective β2 adrenergic receptor (adrenoreceptor) antagonist or beta blocker. ICI binds to the β2 subtype with at least 100 times greater affinity than β1 or β3, the two other known subtypes of the beta adrenoceptor. The compound was developed by Imperial Chemical Industries, which was acquired by AkzoNobel in 2008.
GSK-189,254 is a potent and selective H3 histamine receptor inverse agonist developed by GlaxoSmithKline. It has subnanomolar affinity for the H3 receptor (Ki = 0.2nM) and selectivity of over 10,000x for H3 over other histamine receptor subtypes. Animal studies have shown it to possess not only stimulant and nootropic effects, but also analgesic action suggesting a role for H3 receptors in pain processing in the spinal cord. GSK-189,254 and several other related drugs are currently being investigated as a treatment for Alzheimer's disease and other forms of dementia, as well as possible use in the treatment of conditions such as narcolepsy, or neuropathic pain which do not respond well to conventional analgesic drugs.
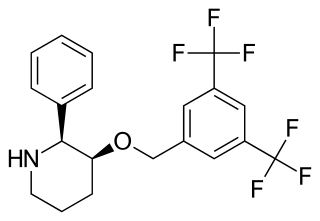
L-733,060 is a drug developed by Merck which acts as an orally active, non-peptide, selective antagonist for the NK1 receptor, binding with a Ki of 0.08 nM. Only one enantiomer is active which has made it the subject of several asymmetric synthesis efforts.
The alpha-3 beta-4 nicotinic receptor, also known as the α3β4 receptor and the ganglion-type nicotinic receptor, is a type of nicotinic acetylcholine receptor, consisting of α3 and β4 subunits. It is located in the autonomic ganglia and adrenal medulla, where activation yields post- and/or presynaptic excitation, mainly by increased Na+ and K+ permeability.
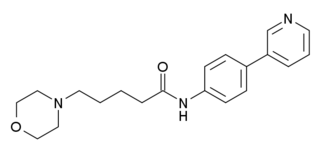
WAY-317538 (SEN-12333) is a drug that acts as a potent and selective full agonist for the α7 subtype of neural nicotinic acetylcholine receptors. It was not the most potent compound in the series, but was selected for further development on the basis of its high selectivity over related receptors, ease of synthesis, and good in vivo properties including high oral bioavailability and good brain penetration. It has nootropic and neuroprotective effects in animal studies, and is being investigated as a potential treatment for neurodegenerative and neurocognitive conditions including Alzheimer's disease and schizophrenia.
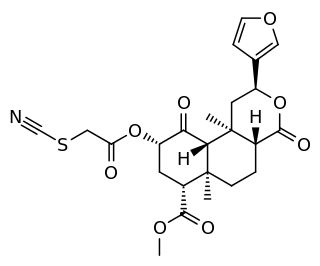
RB-64 is a semi-synthetic derivative of salvinorin A. It is an irreversible agonist, with a reactive thiocyanate group that forms a bond to the κ-opioid receptor (KOR), resulting in very high potency. It is functionally selective, activating G proteins more potently than β-arrestin-2. RB-64 has a bias factor of up to 96 and is analgesic with fewer of the side-effects associated with unbiased KOR agonists. The analgesia is long-lasting. Compared with unbiased agonists, RB-64 evokes considerably less receptor internalization.
Cerebrolysin is a mixture of enzymatically treated peptides derived from pig brain whose constituents can include brain-derived neurotrophic factor (BDNF), glial cell line-derived neurotrophic factor (GDNF), nerve growth factor (NGF), and ciliary neurotrophic factor (CNTF).

Acetothiolutamide is a selective androgen receptor modulator (SARM) derived from the nonsteroidal antiandrogen bicalutamide that was described in 2002 and was one of the first SARMs to be discovered and developed. It is a high-affinity, selective ligand of the androgen receptor (AR), where it acts as a full agonist in vitro, and has in vitro potency comparable to that of testosterone. However, in vivo, acetothiolutamide displayed overall negligible androgenic effects, though significant anabolic effects were observed at high doses. In addition, notable antiandrogen effects were observed in castrated male rats treated with testosterone propionate. The discrepancy between the in vitro and in vivo actions of acetothiolutamide was determined to be related to rapid plasma clearance and extensive hepatic metabolism into a variety of metabolites with differing pharmacological activity, including AR partial agonism and antagonism. In accordance with its poor metabolic stability, acetothiolutamide is not orally bioavailable, and shows activity only via injected routes such as subcutaneous and intravenous.

JNJ-18038683 is a potent and selective antagonist of the 5HT7 serotonin receptor discovered by Johnson & Johnson. It has nootropic and antidepressant effects in both animal and human studies and has progressed to Phase II trials as an adjunctive treatment for improving cognition and mood in stable bipolar disorder; it has been found to reduce REM sleep (the lightest stage of sleep, elevated in depression) in humans and block circadian rhythm phase-shift advances in mice.
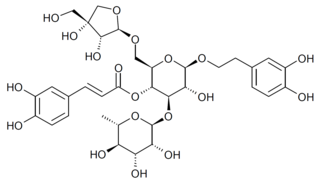
Forsythoside B is a natural product from the phenylpropanoid glycoside group, which is found in a number of plant species such as Marrubium alysson, Phlomis armeniaca, Scutellaria salviifolia, Phlomoides tuberosa, Phlomoides rotata, Pedicularis longiflora and Teucrium chamaedrys, several of which are used in Chinese traditional medicine in preparations such as Shuanghuanglian (双黄连). It acts as an inhibitor of inflammatory mediators such as TNF-alpha, IL-6, IκB and NF-κB, as well as the temperature sensitive channel TRPV3, but also activates the RhoA/ROCK signaling pathway which can cause hypersensitivity reactions when it is injected intravenously.
