List of biophysically important macromolecular crystal structures
Last updated
Nucleosome X-ray crystal structure
Crystal structures of protein and nucleic acid molecules and their complexes are central to the practice of most parts of biophysics, and have shaped much of what we understand scientifically at the atomic-detail level of biology. Their importance is underlined by the United Nations declaring 2014 as the International Year of Crystallography, as the 100th anniversary of Max von Laue's 1914 Nobel Prize for discovering the diffraction of X-rays by crystals. This chronological list of biophysically notable protein and nucleic acid structures is loosely based on a review in the Biophysical Journal.[1] The list includes all the first dozen distinct structures, those that broke new ground in subject or method, and those that became model systems for work in future biophysical areas of research.
1958–Myoglobin was the very first crystal structure of a protein molecule.[2] Myoglobin cradles an iron-containing heme group that reversibly binds oxygen for use in powering muscle fibers, and those first crystals were of myoglobin from the sperm whale, whose muscles need copious oxygen storage for deep dives. The myoglobin 3-dimensional structure is made up of 8 alpha-helices, and the crystal structure showed that their conformation was right-handed and very closely matched the geometry proposed by Linus Pauling, with 3.6 residues per turn and backbone hydrogen bonds from the peptide NH of one residue to the peptide CO of residue i+4. Myoglobin is a model system for many types of biophysical studies,[3] especially involving the binding process of small ligands such as oxygen and carbon monoxide.
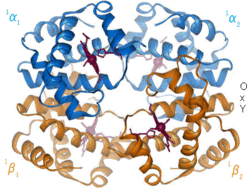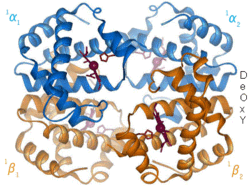
1960– The hemoglobin crystal structure[4] showed a tetramer of two related chain types and was solved at much lower resolution than the monomeric myoglobin, but it clearly had the same basic 8-helix architecture (now called the "globin fold"). Further hemoglobin crystal structures at higher resolution (PDB 1MHB, 1DHB) soon showed the coupled change of both local and quaternary conformation between the oxy and deoxy states of hemoglobin,[5] which explains the cooperativity of oxygen binding in the blood and the allosteric effect of factors such as pH and DPG. For decades hemoglobin was the primary teaching example for the concept of allostery, as well as being an intensive focus of research and discussion on allostery. In 1909, hemoglobin crystals from >100 species were used to relate taxonomy to molecular properties.[6] That book was cited by Perutz in the 1938 report[7] of horse hemoglobin crystals that began his long saga to solve the crystal structure. Hemoglobin crystals are pleochroic— dark red in two directions and pale red in the third[6]— because of the orientation of the hemes, and the bright Soret band of the heme porphyrin groups is used in spectroscopic analysis of hemoglobin ligand binding.
HEW lysozyme ribbon
Hen-egg-white lysozyme
1965– Hen-egg-white lysozyme (PDB file 1lyz).[8] was the first crystal structure of an enzyme (it cleaves small carbohydrates into simple sugars), used for early studies of enzyme mechanism.[9] It contained beta sheet (antiparallel) as well as helices, and was also the first macromolecular structure to have its atomic coordinates refined (in real space).[10] The starting material for preparation can be bought at the grocery store, and hen-egg lysozyme crystallizes very readily in many different space groups; it is the favorite test case for new crystallographic experiments and instruments. Recent examples are nanocrystals of lysozyme for free-electron laser data collection[11] and microcrystals for micro electron diffraction.[12]
Ribonuclease A ribbon drawing
Ribonuclease
1967–Ribonuclease A (PDB file 2RSA)[13] is an RNA-cleaving enzyme stabilized by 4 disulfide bonds. It was used in Anfinsen's seminal research on protein folding which led to the concept that a protein's 3-dimensional structure was determined by its amino-acid sequence. Ribonuclease S, the cleaved, two-component form studied by Fred Richards, was also enzymatically active, had a nearly identical crystal structure (PDB file 1RNS),[14] and was shown to be catalytically active even in the crystal,[15] helping dispel doubts about the relevance of protein crystal structures to biological function.
Two domains of elastase
Serine proteases
1967– The serine proteases are a historically very important group of enzyme structures, because collectively they illuminated catalytic mechanism (in their case, by the Ser-His-Asp "catalytic triad"), the basis of differing substrate specificities, and the activation mechanism by which a controlled enzymatic cleavage buries the new chain end to properly rearrange the active site.[16] The early crystal structures included chymotrypsin (PDB file 2CHA),[17]chymotrypsinogen (PDB file 1CHG),[18]trypsin (PDB file 1PTN),[19] and elastase (PDB file 1EST).[20] They also were the first protein structures that showed two near-identical domains, presumably related by gene duplication. One reason for their wide use as textbook and classroom examples was the insertion-code numbering system, which made Ser195 and His57 consistent and memorable despite the protein-specific sequence differences.[citation needed]
1969–Carboxypeptidase A is a zinc metalloprotease. Its crystal structure (PDB file 1CPA)[21] showed the first parallel beta structure: a large, twisted, central sheet of 8 strands with the active-site Zn located at the C-terminal end of the middle strands and the sheet flanked on both sides with alpha helices. It is an exopeptidase that cleaves peptides or proteins from the carboxy-terminal end rather than internal to the sequence. Later a small protein inhibitor of carboxypeptidase was solved (PDB file 4CPA)[22] that mechanically stops the catalysis by presenting its C-terminal end just sticking out from between a ring of disulfide bonds with tight structure behind it, preventing the enzyme from sucking in the chain past the first residue.
Subtilisin ribbon
Subtilisin
1969–Subtilisin (PDB file 1sbt [23]) was a second type of serine protease with a near-identical active site to the trypsin family of enzymes, but with a completely different overall fold. This gave the first view of convergent evolution at the atomic level. Later, an intensive mutational study on subtilisin documented the effects of all 19 other amino acids at each individual position.[24]
1970– Basic pancreatic trypsin inhibitor, or BPTI (PDB file 2pti[25]), is a small, very stable protein that has been a highly productive model system for study of super-tight binding, disulfide bond (SS) formation, protein folding, molecular stability by amino-acid mutations or hydrogen-deuterium exchange, and fast local dynamics by NMR. Biologically, BPTI binds and inhibits trypsin while stored in the pancreas, allowing activation of protein digestion only after trypsin is released into the stomach.
Rubredoxin with nn-heme iron
Rubredoxin
1970–Rubredoxin (PDB file 2rxn[26]) was the first redox structure solved, a minimalist protein with the iron bound by 4 Cys sidechains from 2 loops at the top of β hairpins. It diffracted to 1.2Å, enabling the first reciprocal-space refinement of a protein (4,5rxn[27]). (NB: note that 4rxn was done without geometry restraints.) Archaeal rubredoxins account for many of the highest-resolution small structures in the PDB.
1971–Insulin (PDB file 1INS)[28] is a hormone central to the metabolism of sugar and fat storage, and important in human diseases such as obesity and diabetes. It is biophysically notable for its Zn binding, its equilibrium between monomer, dimer, and hexamer states, its ability to form crystals in vivo, and its synthesis as a longer "pro" form which is then cleaved to fold up as the active 2-chain, SS-linked monomer. Insulin was a success of NASA's crystal-growth program on the Space Shuttle, producing bulk preparations of very uniform tiny crystals for controlled dosage.
1985–Photosynthetic reaction center: Pairs of bacteriochlorophylls (green) inside the membrane capture energy from sunlight, then traveling by many steps to become available at the heme groups (red) in the cytochrome-C module at the top. This was first crystal structure solved for a membrane protein, a milestone recognized by a Nobel Prize to Hartmut Michel, Hans Deisenhofer, and Robert Huber.
1998–DNA topoisomerases perform the biologically important and necessary job of untangling DNA strands or helices that get entwined with each other or twisted too tightly during normal cellular processes such as the transcription of genetic information.
2009– The vault particle is an intriguing new discovery of a large hollow particle common in cells, with several different suggestions for its possible biological function. The crystal structures (PDB files 2zuo, 2zv4, 2zv5[29] and 4hl8[30]) show that each half of the vault is made up of 39 copies of a long 12-domain protein that swirl together to form the enclosure. Disorder at the very top and bottom ends suggests openings for possible access to the interior of the vault.
↑ Kendrew JC, Bodo G, Dintzis HM, Parrish RG, Wyckoff H, Phillips DC (1958). "A Three-Dimensional Model of the Myoglobin Molecule Obtained by X-Ray Analysis". Nature. 181 (4610): 662–666. Bibcode:1958Natur.181..662K. doi:10.1038/181662a0. PMID13517261.
↑ Perutz MF, Rossmann MG, Cullis AF, Muirhead H, North AC (1960). "Structure of haemoglobin: A three-dimensional Fourier synthesis at 5.5-Å resolution, obtained by X-ray analysis". Nature. 185 (4711): 416–422. Bibcode:1960Natur.185..416P. doi:10.1038/185416a0. PMID18990801. S2CID4208282.
↑ Warshel A, Levitt M (1976). "Theoretical studies of enzymic reactions: Dielectric, electrostatic, and steric stabilization of the carbonium ion in the reaction of lysozyme". J Mol Biol. 103 (2): 227–49. doi:10.1016/0022-2836(76)90311-9. PMID985660.
↑ Diamond R (1974). "Real-space refinement of the structure of hen egg-white lysozyme". Journal of Molecular Biology. 82 (3): 371–374. doi:10.1016/0022-2836(74)90598-1. PMID4856347.
↑ Freer ST, Kraut J, Robertus JD, Tonle H, Wright HT (1970). "Chymotrypsinogen: 2.5-Å crystal structure, comparison with α-chymotrypsin, and implications for zymogen activation". Biochemistry. 9 (9): 1997–2009. doi:10.1021/bi00811a022. PMID5442169.
↑ Fehlhammer H, Bode W (1975). "The refined crystal structure of bovine beta-trypsin at 1.8Å resolution, II. Crystallographic refinement, calcium binding site and active site at pH 7.0". Journal of Molecular Biology. 98 (4): 693–697. doi:10.1016/s0022-2836(75)80005-2. PMID512.
↑ Sawyer L, Shotton DM, Campbell JW, Ladner RC (1978). "The atomic structure of crystalline porcine pancreatic elastase at 2.5Å resolution: comparison with the structure of alpha-chymotrypsin". Journal of Molecular Biology. 118 (2): 137–208. doi:10.1016/0022-2836(78)90412-6. PMID628010.
↑ Lipscomb WN; Hartsuck JA; Reeke GN; Quiocho FA; Bethge PH; Ludwif=g ML; Steitz TA; Muirhead H; Coppola JC (1969). "The structure of carboxypeptidase A, VII. The 2.0-Å resolution studies of the enzyme and of its complex with glycyltyrosine, and mechanistic deductions". Brookhaven Symposia in Biology. 21 (1): 24–90. PMID5719196.
↑ Rees DC, Lipscomb WN (1982). "Refined crystal structure of the potato inhibitor complex of carboxypeptidase A at 2.5 A resolution". Journal of Molecular Biology. 160 (3): 475–498. doi:10.1016/0022-2836(82)90309-6. PMID7154070.
↑ Alden RA, Birktoft JJ, Kraut J, Robertus JD, Wright CS (1971). "Atomic coordinates for subtilisin BPN' (or novo)". Biochem Biophys Res Commun. 45 (2): 337–44. doi:10.1016/0006-291X(71)90823-0. PMID5160720.
↑ Huber R, Kukla D, Ruhmann A, Epp O, Formanek H (1970). "The basic trypsin inhibitor of bovine pancreas. I. Structure analysis and conformation of the polypeptide chain". Naturwissenschaften. 57 (5304): 389–392. Bibcode:1971Natur.231..506B. doi:10.1038/231506a0. PMID4932997. S2CID4158731.
↑ Herriott JR, Sieker LC, Jensen LH, Lovenberg W (1970). "Structure of rubredoxin: An X-ray study to 2.5Å resolution". Journal of Molecular Biology. 50 (2): 391–402. doi:10.1016/0022-2836(70)90200-7. PMID5476919.
↑ Watenpaugh KD, Sieker LC, Jensen LH (1980). "Crystallographic refinement of rubredoxin at 1.2Å resolution". Journal of Molecular Biology. 138 (3): 615–633. doi:10.1016/s0022-2836(80)80020-9. PMID7411618.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.