
Coenzyme A (CoA, SHCoA, CoASH) is a coenzyme, notable for its role in the synthesis and oxidation of fatty acids, and the oxidation of pyruvate in the citric acid cycle. All genomes sequenced to date encode enzymes that use coenzyme A as a substrate, and around 4% of cellular enzymes use it (or a thioester) as a substrate. In humans, CoA biosynthesis requires cysteine, pantothenate (vitamin B5), and adenosine triphosphate (ATP).
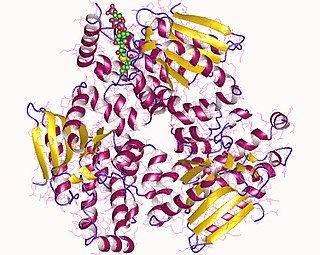
Enoyl-CoA-(∆) isomerase (EC 5.3.3.8, also known as dodecenoyl-CoA- isomerase, 3,2-trans-enoyl-CoA isomerase, ∆3 ,∆2 -enoyl-CoA isomerase, or acetylene-allene isomerase, is an enzyme that catalyzes the conversion of cis- or trans-double bonds of coenzyme A bound fatty acids at gamma-carbon to trans double bonds at beta-carbon as below:
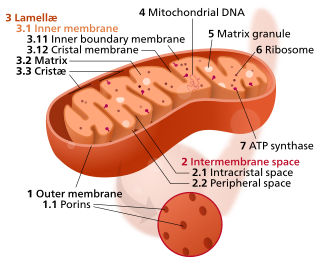
In the mitochondrion, the matrix is the space within the inner membrane. The word "matrix" stems from the fact that this space is viscous, compared to the relatively aqueous cytoplasm. The mitochondrial matrix contains the mitochondrial DNA, ribosomes, soluble enzymes, small organic molecules, nucleotide cofactors, and inorganic ions.[1] The enzymes in the matrix facilitate reactions responsible for the production of ATP, such as the citric acid cycle, oxidative phosphorylation, oxidation of pyruvate, and the beta oxidation of fatty acids.
In biochemistry and metabolism, beta oxidation (also β-oxidation) is the catabolic process by which fatty acid molecules are broken down in the cytosol in prokaryotes and in the mitochondria in eukaryotes to generate acetyl-CoA. Acetyl-CoA enters the citric acid cycle, generating NADH and FADH2, which are electron carriers used in the electron transport chain. It is named as such because the beta carbon of the fatty acid chain undergoes oxidation and is converted to a carbonyl group to start the cycle all over again. Beta-oxidation is primarily facilitated by the mitochondrial trifunctional protein, an enzyme complex associated with the inner mitochondrial membrane, although very long chain fatty acids are oxidized in peroxisomes.
Numerous genetic disorders are caused by errors in fatty acid metabolism. These disorders may be described as fatty oxidation disorders or as a lipid storage disorders, and are any one of several inborn errors of metabolism that result from enzyme defects affecting the ability of the body to oxidize fatty acids in order to produce energy within muscles, liver, and other cell types.

ACADM is a gene that provides instructions for making an enzyme called acyl-coenzyme A dehydrogenase that is important for breaking down (degrading) a certain group of fats called medium-chain fatty acids.
Acyl-CoA dehydrogenases (ACADs) are a class of enzymes that function to catalyze the initial step in each cycle of fatty acid β-oxidation in the mitochondria of cells. Their action results in the introduction of a trans double-bond between C2 (α) and C3 (β) of the acyl-CoA thioester substrate. Flavin adenine dinucleotide (FAD) is a required co-factor in addition to the presence of an active site glutamate in order for the enzyme to function.
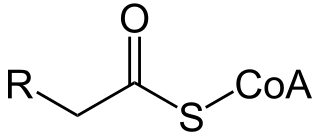
Acyl-CoA is a group of coenzymes that metabolize fatty acids. Acyl-CoA's are susceptible to beta oxidation, forming, ultimately, acetyl-CoA. The acetyl-CoA enters the citric acid cycle, eventually forming several equivalents of ATP. In this way, fats are converted to ATP, the universal biochemical energy carrier.

Electron-transferring-flavoprotein dehydrogenase is an enzyme that transfers electrons from electron-transferring flavoprotein in the mitochondrial matrix, to the ubiquinone pool in the inner mitochondrial membrane. It is part of the electron transport chain. The enzyme is found in both prokaryotes and eukaryotes and contains a flavin and FE-S cluster. In humans, it is encoded by the ETFDH gene. Deficiency in ETF dehydrogenase causes the human genetic disease multiple acyl-CoA dehydrogenase deficiency.
In enzymology, sarcosine dehydrogenase (EC 1.5.8.3) is a mitochondrial enzyme that catalyzes the chemical reaction N-demethylation of sarcosine to give glycine. This enzyme belongs to the family of oxidoreductases, specifically those acting on the CH-NH group of donor with other acceptors. The systematic name of this enzyme class is sarcosine:acceptor oxidoreductase (demethylating). Other names in common use include sarcosine N-demethylase, monomethylglycine dehydrogenase, and sarcosine:(acceptor) oxidoreductase (demethylating). Sarcosine dehydrogenase is closely related to dimethylglycine dehydrogenase, which catalyzes the demethylation reaction of dimethylglycine to sarcosine. Both sarcosine dehydrogenase and dimethylglycine dehydrogenase use FAD as a cofactor. Sarcosine dehydrogenase is linked by electron-transferring flavoprotein (ETF) to the respiratory redox chain. The general chemical reaction catalyzed by sarcosine dehydrogenase is:

In enzymology, a 3-hydroxyacyl-CoA dehydrogenase (EC 1.1.1.35) is an enzyme that catalyzes the chemical reaction

In enzymology, an acyl-CoA dehydrogenase (NADP+) (EC 1.3.1.8) is an enzyme that catalyzes the chemical reaction

The human ETFA gene encodes the Electron-transfer-flavoprotein, alpha subunit, also known as ETF-α. Together with Electron-transfer-flavoprotein, beta subunit, encoded by the 'ETFB' gene, it forms the heterodimeric electron transfer flavoprotein (ETF). The native ETF protein contains one molecule of FAD and one molecule of AMP, respectively.

The human ETFB gene encodes the Electron-transfer-flavoprotein, beta subunit, also known as ETF-β. Together with Electron-transfer-flavoprotein, alpha subunit, encoded by the 'ETFA' gene, it forms the heterodimeric Electron transfer flavoprotein (ETF). The native ETF protein contains one molecule of FAD and one molecule of AMP, respectively.

Electron transfer flavoprotein-ubiquinone oxidoreductase, mitochondrial is an enzyme that in humans is encoded by the ETFDH gene. This gene encodes a component of the electron-transfer system in mitochondria and is essential for electron transfer from a number of mitochondrial flavin-containing dehydrogenases to the main respiratory chain.
A broad classification for genetic disorders that result from an inability of the body to produce or utilize an enzyme or transport protein that is required to oxidize fatty acids. They are an inborn error of lipid metabolism, and when it affects the muscles also a metabolic myopathy.

Short-chain acyl-CoA dehydrogenase is an enzyme with systematic name short-chain acyl-CoA:electron-transfer flavoprotein 2,3-oxidoreductase. This enzyme catalyses the following chemical reaction
Long-chain acyl-CoA dehydrogenase is an enzyme with systematic name long-chain acyl-CoA:electron-transfer flavoprotein 2,3-oxidoreductase. This enzyme catalyses the following chemical reaction

Very-long-chain acyl-CoA dehydrogenase is an enzyme with systematic name very-long-chain acyl-CoA:electron-transfer flavoprotein 2,3-oxidoreductase. This enzyme catalyses the following chemical reaction

NADH:ubiquinone reductase (non-electrogenic) (EC 1.6.5.9, NDH-2, ubiquinone reductase, coenzyme Q reductase, dihydronicotinamide adenine dinucleotide-coenzyme Q reductase, DPNH-coenzyme Q reductase, DPNH-ubiquinone reductase, NADH-coenzyme Q oxidoreductase, NADH-coenzyme Q reductase, NADH-CoQ oxidoreductase, NADH-CoQ reductase) is an enzyme with systematic name NADH:ubiquinone oxidoreductase. This enzyme catalyses the following chemical reaction:

