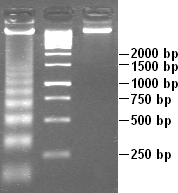
A molecular-weight size marker in the form of a 1kb DNA ladder in the rightmost lane, used in gel electrophoresis. Gel conditions are 1% agarose, 3 volt/cm, and ethidium bromide stain.
A molecular-weight size marker, also referred to as a protein ladder, DNA ladder, or RNA ladder, is a set of standards that are used to identify the approximate size of a molecule run on a gel during electrophoresis, using the principle that molecular weight is inversely proportional to migration rate through a gel matrix. Therefore, when used in gel electrophoresis, markers effectively provide a logarithmic scale by which to estimate the size of the other fragments (providing the fragment sizes of the marker are known).
Protein, DNA, and RNA markers with pre-determined fragment sizes and concentrations are commercially available. These can be run in either agarose or polyacrylamide gels. The markers are loaded in lanes adjacent to sample lanes before the commencement of the run.
DNA markers
Electrophoresed gel with DNA ladders of varying lengths in left lane and middle lane. Fragments sizes are marked on the right, in base pairs.
Development
Although the concept of molecular-weight markers has been retained, techniques of development have varied throughout the years. New inventions of molecular-weight markers are distributed in kits specific to the marker's type.
An early problem in the development of markers was achieving high resolution throughout the entire length of the marker.[1] Depending on the running conditions of gel electrophoresis, fragments may have been compressed, disrupting clarity. To address this issue, a kit for Southern Blot analysis was developed in 1990, providing the first marker to combine target DNA and probe DNA. This technique took advantage of logarithmic spacing, and could be used to identify target bands ranging over a length of 20,000 nucleotides.[2]
Design
There are two common methods in which to construct a DNA molecular-weight size marker.[3] One such method employs the technique of partial ligation.[3] DNA ligation is the process by which linear DNA pieces are connected to each other via covalent bonds; more specifically, these bonds are phosphodiester bonds.[4] Here, a 100bp duplex DNA piece is partially ligated. The consequence of this is that dimers of 200bp, trimers of 300bp, tetramers of 400bp, pentamers of 500bp, etc. will form. Additionally, a portion of the 100bp dsDNA will remain. As a result, a DNA "ladder" composed of DNA pieces of known molecular mass is created on the gel.[3]
The second method employs the use of restriction enzymes and a recognized DNA sequence.[3] The DNA is digested by a particular restriction enzyme, resulting in DNA pieces of varying molecular masses. One of the advantages of this method is that more marker can readily be created simply by digesting more of the known DNA.[3] On the other hand, the size of the DNA pieces are based on the sites where the restriction enzyme cuts. This makes it more difficult to control the size of the fragments in the marker.[5]
More recently, another method for constructing DNA molecular-weight size markers is being employed by laboratories. This strategy involves the use of Polymerase Chain Reaction (PCR).[5] This is achieved one or two ways: 1) a DNA target is amplified at the same time via primer sets, or 2) different DNA targets are amplified independently via particular primers.[5]
Effects of gel conditions
As with experimental samples, the conditions of the gel can affect the molecular-weight size marker that runs alongside them. Factors such as buffer, charge/voltage, and concentration of gel can affect the mobility and/or appearance of your marker/ladder/standard. These elements need to be taken into consideration when selecting a marker and when analyzing the final results on a gel.
1.2% agarose gel showing two different DNA ladders dyed with GelRed stain
Buffers
Buffers act to 1) establish pH, and 2) provide ions to support conductivity. In DNA electrophoresis, the TAE (Tris-acetate-EDTA) and TBE (Tris-borate-EDTA) are the usual buffers of choice.[6] TBE buffer is preferred for small DNA pieces, whereas TAE is better suited for fragments greater than 1500 base pairs. In terms of buffering capacity, TAE is lower when compared to TBE; this generally results in slower mobility of the DNA. TBE is also capable of better resolution.[7]
Water cannot act as a substitute for one of these buffers, as the DNA will not migrate along the gel.[6] Furthermore, using water instead of buffer will result in the gel melting.[8]
Charge/Voltage
In terms of voltage, the recommended range is between 4 and 10 V/cm (i.e., volts/cm).[8] Agarose gels are usually run at a voltage of 5 V/cm.[3][6] The distance unit, cm, refers to the distance between the electrodes (i.e., the anode and the cathode) and not the length of the gel itself.[3][6]
Voltages too far below or above this range will affect the mobility and the resolution of the bands. Low voltages will decrease the mobility and will cause the bands to broaden. On the other hand, high voltages will decrease the resolution of the bands. This is largely due to the fact that voltages that are too high can cause the gel to overheat, and even melt.[8]
Concentration
Agarose concentration must be taken into account when selecting a marker. The gel percentage effects the migration of the DNA.[3][6] Generally, the higher the gel concentration, the slower the rate at which the DNA will move through the gel. This is in addition to the role molecular weight plays in the migration of a DNA marker or sample, that is to say, that the higher the molecular weight, the slower the DNA will migrate.[3][6]
Gel concentration also affects the ability to visualize the bands run out on the gel. Smaller bands are better resolved on a higher percentage gel, whereas increased molecular-weight bands are more easily visualized on a lower percentage gel.[6]
Protein markers
Development
Previously, protein markers had been developed using a variety of whole proteins. The development of a kit including a molecular-weight size marker based on protein fragments began in 1993. This protein marker, composed of 49 different amino acid sequences, included multidomain proteins, and allowed for the analysis of proteins cleaved at different sites.[9]
Current technique improvements in protein markers involve the use of auto-development. The first auto-developed regularly-weight protein marker was invented in 2012.[10]
Design
Similar to DNA markers, these markers are typically composed of purified proteins whose molecular masses are already known.[3] The list below outlines some of the proteins, as well as the molecular mass, that are commonly used when constructing a protein marker.
Immunoblot with protein ladder in leftmost lane. Fragment sizes are measured in kDa (kilodaltons).
Molecular-weight size markers can be broken up into two categories: molecular weight markers vs. molecular ladder markers.[14] Markers are either stained or unstained, and depending on the circumstance, one may be more appropriate than another. Molecular-weight size markers can also be biochemically altered.[15] The conjugation with biotin is the most common. Molecular-weight size markers are most commonly used in SDS-polyacrylamide gel electrophoresis and western blotting. With all the different types and uses of molecular-weight size markers, it is important to choose the appropriate protein standard. Besides the most common use, as a way to calculate the molecular weight of the samples, other uses include allowing visual evidence of protein migration and transfer efficiency and are sometimes even used for positive control.[16]
MW marker vs Protein Ladders
A molecular weight marker is one type of protein standard. They can either prestained or unstained prior to loading; depending on the type of experiment one may be more advantageous. In either case, they are normally run on the outer lane of a gel, while the sample is loaded in the middle lanes.[14] Molecular markers are different from protein ladders in that they are composed of a mixture of native proteins whose specifications are well categorized but do not correspond to whole numbers.[14] Generally these are much cheaper, but analysis only allows for an approximate value of the electrophoresis separated proteins.[14]
A protein ladder is another type of protein standard. They are almost always stained.[14] Protein ladders differ from molecular markers in that they are composed of a mixture of highly purified proteins whose specifications are known and correspond to whole numbers.[14] Generally protein ladders are composed of 10-12 proteins.[14] At the end of the experiment, after size migration occurs, a single band will represent the size of each protein contained in the ladder.[17] Markers are evenly spaced, and size analysis using these markers allows for a precise value of the protein of interest. In some cases, as a molecular confirmation method, MW markers are run with protein ladders for verification.[14]
Prestained and Unstained Markers
Protein markers can come unstained or prestained, but both have their benefits and disadvantages.[18] Simple visualization of protein separation and transfer is made possible through the use of prestained markers.[18] They are commonly used in both SDS-polyacrylamide gel electrophoresis and western blotting. In SDS-PAGE it allows for the monitoring of protein migration, as the protein bands will separate and can be seen during an electrophoretic run. In western blots, the stained protein standards allow for the visualization protein transfer onto the membrane.[17] However, size determinations are not as accurate with these markers (see Recombinant and Natural Marker section for further explanation).[18]
While the unstained markers allow for more exact size determinations, they cannot be viewed while the gel is running. As such, the gel must be stained in order to visualize the bands.[19]
Recombinant and Natural Markers
Besides stained and unstained markers, protein markers can be thought of in terms of recombinant and natural.[18] Recombinant markers consist of recombinant proteins which have been greatly purified. These markers are designed in such a manner as to highlight particular characteristics.[18] Examples of these characteristics include affinity tags and molecular weights which are uniformly positioned relative to each other.[18]
Natural markers, as the name implies, are a mixture of proteins which happen naturally.[18] Prestained natural markers work well for gel separation visualization. However, these markers tend to bind to the stain in a covalent manner in varying amounts and at various positions.[18] Consequently, the resultant bands may be broader. This is especially true when making comparisons to prestained recombinant markers. Due to this effect, molecular weight determinations are likely to be less accurate with the prestained natural markers.[18]
Biochemically Altered
Protein standards can also be chemically altered. A common alteration is through the use biotin. Biotin has a very high affinity for streptavidin, and therefore, the binding forms a very strong complex. For visualization, a color tag is attached to the streptavidin.[15]
Effects of gel conditions
As with DNA electrophoresis, conditions such as buffers, charge/voltage, and concentration should be taken into account when selecting a protein marker.
Buffers
Buffers can affect the mobility of both the marker and the samples. The pH of the buffer varies with the system used and consequently, each buffer system will have a different effect of the charge of a protein or proteins.[20] In addition, in the case of SDS-PAGE, the binding affinity for SDS can be affected by the buffering system.[20] Even when using the same percentage and type of gel, the same proteins will migrate at different rates depending on the buffer used.[20]
Charge/Voltage
Voltage plays a role in the mobility of proteins on a gel. Proteins will migrate faster at higher voltages. Consequently, the gel running time will be shorter. Conversely, higher voltages can result in greater band diffusion.[20] Also, if the voltage is too high, the temperature in the electrophoresis chamber can become such that the gel begins to melt.[20]
The voltage that a gel should be run at depends on the type of the gel. For some gels, the voltage remains constant throughout the run, whereas, with other gels, the initial voltage is allowed to remain constant for a specified time before it is increased.[20] This second voltage is then used for a specific time frame, after which, it may also be increased.[20]
Concentration
In terms of percentage, gels used for protein electrophoresis can be broken down into single-percentage gels and gradient gels.[18] Single-percentage gels are also referred to as linear gels.[20] For linear gels, the selected percentage usually falls between 7.5% and 20%.[18] Common percentage ranges for gradient gels are 4-15% and 10-20%. Each type of gel has its own advantages.[18] For instance, linear gels are preferred when several proteins have similar molecular weights; better separation between these proteins will be displayed by a linear gel.[18] On the other hand, gradient gels are a better choice when the samples of interest contain proteins of vastly different molecular weights or that cover a large range of molecular weights.[18][20]
RNA markers
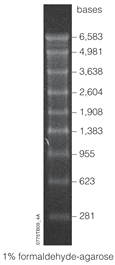
Lane of electrophoresed agarose gel showing an RNA ladder with bands at a range of 281-6583 base pairs
Development
RNA ladders composed of RNA molecular-weight size markers were initially developed by using the synthetic circle method[21] to produce different-sized markers. This technique was improved upon by inventor Eric T. Kool to use circular DNA vectors as a method for producing RNA molecular-weight size markers. As referred to as the rolling circle method, the improvements of this technique stems from its efficiency in synthesizing RNA oligonucleotides. From the circular DNA template, single-stranded RNA varying in length from 4-1500 bp can be produced without the need for primers and by recycling nucleotide triphosphate. DNA can also be synthesized from the circular template, adding to this technique's versatility. In comparison to runoff transcription, the synthetic circle method produces RNA oligonucleotides without the runoff. In comparison to PCR, the synthetic circle method produces RNA oligonucleotides without the need for polymerase nor a thermal cycler. This method is also cost-efficient in its ability to synthesize grand amounts of product at a lower error rate than machine synthesizers.[21]
Design
The RNA markers consist of RNA transcripts of various incrementing lengths. For example, the Lonza 0.5-9 kbp marker[22] has bands marking 0.5, 1, 1.5, 2, 2.5, 3, 4, 5, 6, and 9 kilobase pairs. Markers are dissolved in a storage buffer, such as EDTA, and can have a shelf life of up to 2 years when stored at -80°C. To use the marker, such as for northern blot analysis, it is first thawed, and then stained so that it is detectable on a gel electrophoresis. One of the most common dyes used for markers is ethidium bromide.
The range of a particular marker refers to variety of bands it can map. A "high" range refers to relatively large fragments (measured in kb) while a "low" range refers to markers that distinguish between small fragments (measured in bp). Some markers can even be described as "ultra-low range",[16] but even more precise is the microRNA marker. A microRNA marker can be used to measure RNA fragments within a dozen nucleotides, such as the 17-25 nt microRNA marker.[23]
Use
At equivalent molecular weights, RNA will migrate faster than DNA. However, both RNA and DNA have a negative linearslope between their migration distance and logarithmic molecular weight.[24] That is, samples of less weight are able to migrate a greater distance. This relationship is a consideration when choosing RNA or DNA markers as a standard.
When running RNA markers and RNA samples on a gel, it is important to prevent nuclease contamination, as RNA is very sensitive to ribonuclease (RNase) degradation through catalysis.[25][26] Thus, all materials to be used in the procedure must be taken into consideration. Any glassware that is to come into contact with RNA should be pretreated with diethylpyrocarbonate (DEPC) and plastic materials should be disposable.[25]
Molecular-weight size markers and SDS-PAGE
SDS-PAGE gel using a molecular-weight size standard in the left outer lane
One of the most common uses for molecular-weight size markers is in gel electrophoresis. The purpose of gel electrophoresis is to separate proteins by physical or chemical properties, which include charge, molecular size, and pH.< When separating based on size, the ideal method is SDS-PAGE or polyacrylamide gel electrophoresis and molecular-weight size markers are the appropriate standards to use.
Gels can vary in size. The number of samples to be run will determine the appropriate gel size. All gels are divided into lanes that run parallel through the gel. Each lane will contain a specific sample. Typically, molecular-weight size standards are placed in an outer lane. If a gel has a particularly high number of lanes, then multiple ladders may be placed across the gel for higher clarity.
Proteins and standards are pipetted on the gel in appropriate lanes. Sodium dodecyl sulfate (SDS) interacts with proteins, denaturing them, and giving them a negative charge. Since all proteins have the same charge-to-mass ratio, protein mobility through the gel will solely be based on molecular weight. Once the electric field is turned on, protein migration will initiate. Upon completion, a detection mechanism such as western blotting can be used, which will reveal the presence of bands. Each band represents a specific protein. The distance of travel is solely based on molecular weight; therefore, the molecular weight of each protein can be determined by comparing the distance of an unknown protein to the standard of known molecular weight.[27]
Different uses of molecular-weight size markers
Many kinds of molecular-weight size markers exist, and each possess unique characteristics, lending to their involvement in a number of biological techniques. Selection of a molecular-weight size marker depends upon the marker type (DNA, RNA, or protein) and the length range it offers (e.g. 1kb). Before selecting a molecular-weight size marker, it is important to become familiar with these characteristics and properties. In a particular instance one type may be more appropriate than another. Although specific markers can vary between protocols for a given technique, this section will outline general markers and their roles.
Allozymes
The first type of molecular marker developed and run on gel electrophoresis were allozymes. These markers are used for the detection of protein variation. The word "allozyme" (also known as "alloenzyme") comes from "allelic variants of enzymes."[28] When run on a gel, proteins are separated by size and charge. Although allozymes may seem dated when compared to the other markers available, they are still used today, mainly due to their low cost. One major downside is that since there is only a limited amount available, specificity an issue.[28]
DNA-based markers (1960s)
Although allozymes can detect variations in DNA, it is by an indirect method and not very accurate. DNA-based markers were developed in the 1960s.[28] These markers are much more effective at distinguishing between DNA variants. Today these are the most commonly used markers. DNA-based markers work by surveying nucleotides, which can serve a variety of functions, such as detecting differences in nucleotides or even quantifying the number of mutations.[28]
RFLP
Restriction fragment length polymorphism is a technique used to detect variations in homologous DNA.[29] Specific restriction endonucleases are used to digest DNA. The RFLP molecular marker is specific to a single fragment. Along with alleic RFLP markers, a molecular-weight size marker, in this case a DNA marker,[30] is also included on an electorphoresed agarose gel. The DNA marker allows for the size of the restriction fragments to be estimated.
Minisatellites
Similar to RFLP, this technique also uses restriction endonucleases to digest the genomic DNA. Minisatellites are short sequences of tandem repeats, approximately 10-60 base pairs. Minisatellites can be used in DNA footprinting and as regulators of gene control.[28]
PCR-based markers (1980s)
The success of DNA based markers lead to the development of PCR. PCR (polymerase chain reaction) is a DNA amplification technique that can be applied to various types of fragments. Prior to this development, to amplify DNA, it had to be cloned or isolated. Shortly after the discovery of PCR came the idea of using PCR-based markers for gel electrophoresis. These types of markers are based on PCR primers and are categorized as DNA sequence polymorphism.[28]
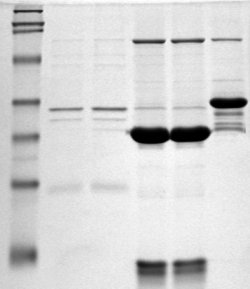
An electrophoresed gel showing PCR products. The leftmost lane represents a DNA ladder with fragments at 100bp intervals.
Microsatellites
Also known as SSR (simple sequence repeats) or STR (short tandem repeats), microsatellites differ from minisatellites in that they are shorter, usually 2-6 base pairs. This property of microsatellites allows for easy isolation. Microsatellites are most commonly used in population genetics. Microsatellites have a high and complex mutation rate, which is their major disadvantage.[28]
AFLP
Amplified fragment length polymorphism is a PCR-based DNA fingerprinting technique. DNA is first digested with endonucleases. The restriction fragments are then ligated together.[31] A molecular marker is then generated when specific fragments are selected for amplification. AFLP markers are run alongside a DNA marker on a gel. A common AFLP DNA marker is 30-330bp long.[32] The fragments of this marker lie at 10bp intervals to increase precision.
RAPD
Random amplified polymorphic DNA is a technique that is conducted similar to AFLP. The difference is that the molecular markers are generated at random.[31] The most common molecular-weight size marker for this technique is the 1kb DNA ladder.[33][34]
DNA sequence polymorphism
Although technically speaking, DNA sequence polymorphism has been going on since the use of RFLP in the 1960s, the analysis has changed significantly over the years. DNA sequence polymorphism uses older techniques like RFLP, but on a larger scale. Sequencing is much faster and more efficient. The analysis is automated, as it uses a technique known as shotgun sequencing. This high-throughput method is commonly used in population genetics.[28]
SNPs
SNPs (single nucleotide polymorphism), are used to detect variations in single nucleotides. The technique is very similar to that of RFLP. SNPs are used frequently for population genetic studies.[35] After amplification through PCR, these small fragments can be visualized using gel electrophoresis, and again DNA markers play a role in determining fragment length.
Polysaccharide analysis by carbohydrate gel electrophoresis
Carbohydrate markers are employed in a technique known as polysaccharide analysis by carbohydrate gel electrophoresis (PACE), which is a measurable separation technique.[36] It allows for the analysis of enzyme hydrolysis products.[36] It has been used in applications such as characterizing enzymes involved in hemicellulose degradation, determining the structure of hemicellulose polysaccharides, and analysis of enzymatic cleavage of cellulose products.[36]
PACE depends on derivitization, which is the conversion of a chemical compound into a derivative.[36][37] Here monosaccharides, oligosaccharides, and polysaccharides are the compounds of interest. They are labeled at their reducing ends with a fluorescent label (i.e. a fluorophore).[36] This derivitization with a fluorophore permits both separation on a gel under the desired circumstances and fluorescence imaging of the gel. In this case, a polyacrylamide gel is used.[36]
As with DNA, RNA, and protein electrophoresis, markers are run alongside the samples of interest in carbohydrate gel electrophoresis.[36] The markers consist of oligosaccharides of known molecular weight. Like the samples of interest, the marker is also derivatized with a fluorophore (usually with 8-aminonaphthalene-1,3,6-trisulfonic acid (ANTS) or 2-aminoacridone).[36]
↑Lonza. "RNA Markers 0.5–9 kbp"(PDF). Document #18123-0807-06. Lonza Rockland Inc. Retrieved 27 November 2013.
↑New England Biolabs. "microRNA Marker". New England Bioloabs. Retrieved 27 November 2013.
↑Wicks, Richard J. (1986). "RNA molecular weight determination by agarose gel electrophoresis using formaldehyde as denaturant: Comparison of rna and dna molecular weight markers". International Journal of Biochemistry. 18 (3): 277–278. doi:10.1016/0020-711x(86)90118-7. PMID2937672.
↑Cammack R, Attwood TK, Campbell PN, Parish HJ, Smith A, Stirling JL, Vella F (2006). "Fluorophore". Oxford Dictionary of Biochemistry and Molecular Biology (Seconded.). Oxford University Press.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.