
Muscular dystrophies (MD) are a genetically and clinically heterogeneous group of rare neuromuscular diseases that cause progressive weakness and breakdown of skeletal muscles over time. The disorders differ as to which muscles are primarily affected, the degree of weakness, how fast they worsen, and when symptoms begin. Some types are also associated with problems in other organs.

A glycogen storage disease is a metabolic disorder caused by a deficiency of an enzyme or transport protein affecting glycogen synthesis, glycogen breakdown, or glucose breakdown, typically in muscles and/or liver cells.

Limb–girdle muscular dystrophy (LGMD) is a genetically heterogeneous group of rare muscular dystrophies that share a set of clinical characteristics. It is characterised by progressive muscle wasting which affects predominantly hip and shoulder muscles. LGMD usually has an autosomal pattern of inheritance. It currently has no known cure or treatment.
Nemaline myopathy is a congenital, often hereditary neuromuscular disorder with many symptoms that can occur such as muscle weakness, hypoventilation, swallowing dysfunction, and impaired speech ability. The severity of these symptoms varies and can change throughout one's life to some extent. The prevalence is estimated at 1 in 50,000 live births. It is the most common non-dystrophic myopathy.
Hereditary inclusion body myopathies (HIBM) are a group of rare genetic disorders which have different symptoms. Generally, they are neuromuscular disorders characterized by muscle weakness developing in young adults. Hereditary inclusion body myopathies comprise both autosomal recessive and autosomal dominant muscle disorders that have a variable expression (phenotype) in individuals, but all share similar structural features in the muscles.

Mitochondrial myopathies are types of myopathies associated with mitochondrial disease. Adenosine triphosphate (ATP), the chemical used to provide energy for the cell, cannot be produced sufficiently by oxidative phosphorylation when the mitochondrion is either damaged or missing necessary enzymes or transport proteins. With ATP production deficient in mitochondria, there is an over-reliance on anaerobic glycolysis which leads to lactic acidosis either at rest or exercise-induced.

Centronuclear myopathies (CNM) are a group of congenital myopathies where cell nuclei are abnormally located in the center of muscle cells instead of their normal location at the periphery.

Congenital muscular dystrophies are autosomal recessively-inherited muscle diseases. They are a group of heterogeneous disorders characterized by muscle weakness which is present at birth and the different changes on muscle biopsy that ranges from myopathic to overtly dystrophic due to the age at which the biopsy takes place.

Emery–Dreifuss muscular dystrophy (EDMD) is a type of muscular dystrophy, a group of heritable diseases that cause progressive impairment of muscles. EDMD affects muscles used for movement, causing atrophy, weakness and contractures. It almost always affects the heart, causing abnormal rhythms, heart failure, or sudden cardiac death. It is rare, affecting 0.39 per 100,000 people. It is named after Alan Eglin H. Emery and Fritz E. Dreifuss.
Congenital myopathy is a very broad term for any muscle disorder present at birth. This defect primarily affects skeletal muscle fibres and causes muscular weakness and/or hypotonia. Congenital myopathies account for one of the top neuromuscular disorders in the world today, comprising approximately 6 in 100,000 live births every year. As a whole, congenital myopathies can be broadly classified as follows:

Bethlem myopathy is predominantly an autosomal dominant myopathy, classified as a congenital form of limb-girdle muscular dystrophy. There are two types of Bethlem myopathy, based on which type of collagen is affected.

Ryanodine receptor 1 (RYR-1) also known as skeletal muscle calcium release channel or skeletal muscle-type ryanodine receptor is one of a class of ryanodine receptors and a protein found primarily in skeletal muscle. In humans, it is encoded by the RYR1 gene.

Central core disease (CCD), also known as central core myopathy, is an autosomal dominant inherited muscle disorder present from birth that negatively affects the skeletal muscles. It was first described by Shy and Magee in 1956. It is characterized by the appearance of the myofibril under the microscope.
Ullrich congenital muscular dystrophy (UCMD) is a form of congenital muscular dystrophy. There are two forms: UCMD1 and UCMD2.

X-linked myotubular myopathy (MTM) is a form of centronuclear myopathy (CNM) associated with mutations in the myotubularin 1 gene. It is found almost always in male infants. It is one of the severest congenital muscle diseases and is characterized by marked muscle weakness, hypotonia and feeding and breathing difficulties.

Congenital fiber type disproportion (CFTD) is an inherited form of myopathy with small type 1 muscle fibers that may occur in a number of neurological disorders. It has a relatively good outcome and follows a stable course. While the exact genetics is unclear, there is an association with mutations in the genes TPM3, ACTA1 and SELENON. It is a rare condition.
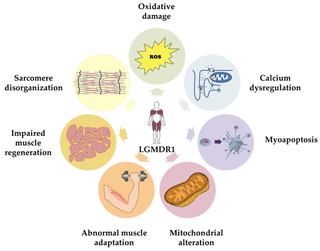
Calpainopathy is the most common type of autosomal recessive limb-girdle muscular dystrophy (LGMD). It preferentially affects the muscles of the hip girdle and shoulder girdle.
Rigid spine syndrome, also known as congenital muscular dystrophy with rigidity of the spine (CMARS), is a rare and often debilitating neuromuscular disorder. It is characterized by progressive muscle stiffness and rigidity, particularly in the spine, which can severely limit mobility and impact quality of life. This condition is typically present from birth or early childhood and tends to worsen over time.

LAMA2 muscular dystrophy (LAMA2-MD) is a genetically determined muscle disease caused by pathogenic mutations in the LAMA2 gene. It is a subtype of a larger group of genetic muscle diseases known collectively as congenital muscular dystrophies. The clinical presentation of LAMA2-MD varies according to the age at presentation. The severe forms present at birth and are known as early onset LAMA2 congenital muscular dystrophy type 1A or MDC1A. The mild forms are known as late onset LAMA2 muscular dystrophy or late onset LAMA2-MD. The nomenclature LGMDR23 can be used interchangeably with late onset LAMA2-MD.