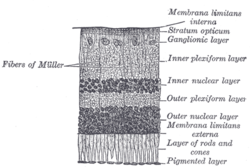
Congenital stationary night blindness (CSNB) is a rare non-progressive retinal disorder. People with CSNB often have difficulty adapting to low light situations due to impaired photoreceptortransmission. These patients may also have reduced visual acuity, myopia, nystagmus, fundus abnormalities, and strabismus.[1][2] CSNB has two forms -- complete, also known as type-1 (CSNB1), and incomplete, also known as type-2 (CSNB2), which are distinguished by the involvement of different retinal pathways. In CSNB1, downstream neurons called bipolar cells are unable to detect neurotransmission from photoreceptor cells. CSNB1 can be caused by mutations in various genes involved in neurotransmitter detection, including NYX. In CSNB2, the photoreceptors themselves have impaired neurotransmission function; this is caused primarily by mutations in the gene CACNA1F, which encodes a voltage-gated calcium channel important for neurotransmitter release. CSNB has been identified in horses and dogs as the result of mutations in TRPM1 (Horse, "LP")[3], GRM6 (Horse, "CSNB2")[4], and LRIT3 (Dog, CSNB)[5].
Congenital stationary night blindness (CSNB) can be inherited in an X-linked, autosomal dominant, or autosomal recessive pattern, depending on the genes involved.
Two forms of CSNB can also affect horses, one linked to the leopard complex of equine coat colors and the other found in certain horse breeds. Both are autosomal recessives.[6][7]
Symptoms and signs
The X-linked varieties of congenital stationary night blindness (CSNB) can be differentiated from the autosomal forms by the presence of myopia, which is typically absent in the autosomal forms. Patients with CSNB often have impaired night vision, myopia, reduced visual acuity, strabismus and nystagmus. Individuals with the complete form of CSNB (CSNB1) have highly impaired rod sensitivity (reduced ~300x) as well as cone dysfunction. Patients with the incomplete form can present with either myopia or hyperopia.[8]
Cause
CSNB is caused by malfunctions in neurotransmission from rod and cone photoreceptors to bipolar cells in the retina.[9] At this first synapse, information from photoreceptors is divided into two channels: ON and OFF. The ON pathway detects light onset, while the OFF pathway detects light offset.[10] The malfunctions in CSNB1 specifically affect the ON pathway, by hindering the ability of ON-type bipolar cells to detect neurotransmitter released from photoreceptors.[9] Rods, which are responsible for low-light vision, make contacts with ON-type bipolar cells only, while, cones, which are responsible for bright-light vision, make contacts with bipolar cells of both ON an OFF subtypes.[11] Because the low-light sensing rods feed only into the ON pathway, individuals with CSNB1 typically have problems with night vision, while vision in well-lit conditions is spared.[9] In CSNB2, release of neurotransmitter from photoreceptors is impaired, leading to involvement of both ON and OFF pathways.[citation needed]
The electroretinogram (ERG) is an important tool for diagnosing CSNB. The ERG a-wave, which reflects the function of the phototransduction cascade in response to a light flashes, is typically normal in CSNB patients, although in some cases phototransduction is also affected, leading to a reduced a-wave. The ERG b-wave, which primarily reflects the function of ON-bipolar cells, is greatly reduced in CSNB2 cases, and completely absent in CSNB1 cases.[9][12]
Genetics
Only three rhodopsin mutations have been found associated with congenital stationary night blindness (CSNB).[13] Two of these mutations are found in the second transmembrane helix of rhodopsin at Gly-90 and Thr-94. Specifically, these mutations are the Gly90Asp [14] and the Thr94Ile, which has been the most recent one reported.[15] The third mutation is Ala292Glu, and it is located in the seventh transmembrane helix, in proximity to the site of retinal attachment at Lys-296.[16] Mutations associated with CSNB affect amino acid residues near the protonated Schiff base (PSB) linkage. They are associated with changes in conformational stability and the protonated status of the PSB nitrogen.[17]
Pathophysiology
CSNB1
The complete form of X-linked congenital stationary night blindness, also known as nyctalopia, is caused by mutations in the NYX gene (Nyctalopin on X-chromosome), which encodes a small leucine-rich repeat (LRR) family protein of unknown function.[18][19] This protein consists of an N-terminal signal peptide and 11 LRRs (LRR1-11) flanked by cysteine-rich LRRs (LRRNT and LRRCT). At the C-terminus of the protein there is a putative GPI anchor site. Although the function of NYX is yet to be fully understood, it is believed to be located extracellularly. A naturally occurring deletion of 85 bases in NYX in some mice leads to the "nob" (no b-wave) phenotype, which is highly similar to that seen in CSNB1 patients.[20] NYX is expressed primarily in the rod and cone cells of the retina. There are currently almost 40 known mutations in NYX associated with CSNB1, Table 1., located throughout the protein. As the function of the nyctalopin protein is unknown, these mutations have not been further characterized. However, many of them are predicted to lead to truncated proteins that, presumably, are non-functional.[citation needed]
LRR: leucine-rich repeat, LRRNT and LRRCT: N- and C-terminal cysteine-rich LRRs.
CSNB2
Figure 1. Schematic structure of CaV1.4 with the domains and subunits labeled.
The incomplete form of X-linked congenital stationary night blindness (CSNB2) is caused by mutations in the CACNA1F gene, which encodes the voltage-gated calcium channel CaV1.4 expressed heavily in retina.[24][25] One of the important properties of this channel is that it inactivates at an extremely low rate. This allows it to produce sustained Ca2+ entry upon depolarization. As photoreceptors depolarize in the absence of light, CaV1.4 channels operate to provide sustained neurotransmitter release upon depolarization.[26] This has been demonstrated in CACNA1F mutant mice that have markedly reduced photoreceptor calcium signals.[27] There are currently 55 mutations in CACNA1F located throughout the channel, Table 2 and Figure 1. While most of these mutations result in truncated and, likely, non-functional channels, it is expected that they prevent the ability of light to hyperpolarize photoreceptors. Of the mutations with known functional consequences, 4 produce channels that are either completely non-functional, and two that result in channels which open at far more hyperpolarized potentials than wild-type. This will result in photoreceptors that continue to release neurotransmitter even after light-induced hyperpolarization.[citation needed]
Table 2. Mutations in CACNA1F associated with CSNB2
Night blindness is a symptom in many patients and diagnosis often occurs through the use of various tests including a electroretinogram to reveal any impairment in the retina "as a whole".[37][38][39] Tests performed can also include a visual field examination, Fundoscopic examination, and slit-lamp microscopy in addition to measurements provided by the electroretinogram (ERG).[40][41][42]
↑Zeitz, Christina; Robson, Anthony G.; Audo, Isabelle (2015-03-01). "Congenital stationary night blindness: An analysis and update of genotype–phenotype correlations and pathogenic mechanisms". Progress in Retinal and Eye Research. 45: 58–110. doi:10.1016/j.preteyeres.2014.09.001. ISSN1350-9462. PMID25307992.
1234Zeitz C, Robson AG, Audo I (March 2015). "Congenital stationary night blindness: an analysis and update of genotype-phenotype correlations and pathogenic mechanisms". Progress in Retinal and Eye Research. 45: 58–110. doi:10.1016/j.preteyeres.2014.09.001. PMID25307992. S2CID45696921.
↑Euler T, Haverkamp S, Schubert T, Baden T (August 2014). "Retinal bipolar cells: elementary building blocks of vision". Nature Reviews. Neuroscience. 15 (8): 507–519. doi:10.1038/nrn3783. PMID25158357. S2CID16309488.
↑Audo I, Robson AG, Holder GE, Moore AT (2008). "The negative ERG: clinical phenotypes and disease mechanisms of inner retinal dysfunction". Survey of Ophthalmology. 53 (1): 16–40. doi:10.1016/j.survophthal.2007.10.010. PMID18191655.
↑N. al-Jandal, G.J. Farrar, A.S. Kiang, M.M. Humphries, N. Bannon, J.B. Findlay, P. Humphries and P.F. Kenna Hum. Mutat. 13 (1999), pp. 75–81.
↑Dryja TP, Berson EL, Rao VR, Oprian DD (July 1993). "Heterozygous missense mutation in the rhodopsin gene as a cause of congenital stationary night blindness". Nature Genetics. 4 (3): 280–3. doi:10.1038/ng0793-280. PMID8358437. S2CID7682929.
123456789101112131415Bech-Hansen NT, Naylor MJ, Maybaum TA, Sparkes RL, Koop B, Birch DG, etal. (November 2000). "Mutations in NYX, encoding the leucine-rich proteoglycan nyctalopin, cause X-linked complete congenital stationary night blindness". Nature Genetics. 26 (3): 319–323. doi:10.1038/81619. PMID11062471. S2CID10223880.
1234567891011121314151617Pusch CM, Zeitz C, Brandau O, Pesch K, Achatz H, Feil S, etal. (November 2000). "The complete form of X-linked congenital stationary night blindness is caused by mutations in a gene encoding a leucine-rich repeat protein". Nature Genetics. 26 (3): 324–327. doi:10.1038/81627. PMID11062472. S2CID42428370.
123456789Zito I, Allen LE, Patel RJ, Meindl A, Bradshaw K, Yates JR, etal. (February 2003). "Mutations in the CACNA1F and NYX genes in British CSNBX families". Human Mutation. 21 (2): 169. doi:10.1002/humu.9106. PMID12552565. S2CID13143864.
1234567Zeitz C, Minotti R, Feil S, Mátyás G, Cremers FP, Hoyng CB, Berger W (March 2005). "Novel mutations in CACNA1F and NYX in Dutch families with X-linked congenital stationary night blindness". Molecular Vision. 11: 179–183. PMID15761389.
1234567891011121314151617181920Boycott KM, Maybaum TA, Naylor MJ, Weleber RG, Robitaille J, Miyake Y, etal. (February 2001). "A summary of 20 CACNA1F mutations identified in 36 families with incomplete X-linked congenital stationary night blindness, and characterization of splice variants". Human Genetics. 108 (2): 91–97. doi:10.1007/s004390100461. PMID11281458. S2CID2844173.
12345Nakamura M, Ito S, Terasaki H, Miyake Y (June 2001). "Novel CACNA1F mutations in Japanese patients with incomplete congenital stationary night blindness". Investigative Ophthalmology & Visual Science. 42 (7): 1610–1616. PMID11381068.
↑Nakamura M, Ito S, Piao CH, Terasaki H, Miyake Y (July 2003). "Retinal and optic disc atrophy associated with a CACNA1F mutation in a Japanese family". Archives of Ophthalmology. 121 (7): 1028–1033. doi:10.1001/archopht.121.7.1028. PMID12860808.
12Hoda JC, Zaghetto F, Singh A, Koschak A, Striessnig J (March 2006). "Effects of congenital stationary night blindness type 2 mutations R508Q and L1364H on Cav1.4 L-type Ca2+ channel function and expression". Journal of Neurochemistry. 96 (6): 1648–1658. doi:10.1111/j.1471-4159.2006.03678.x. PMID16476079. S2CID25987619.
↑Jacobi FK, Hamel CP, Arnaud B, Blin N, Broghammer M, Jacobi PC, etal. (May 2003). "A novel CACNA1F mutation in a french family with the incomplete type of X-linked congenital stationary night blindness". American Journal of Ophthalmology. 135 (5): 733–736. doi:10.1016/S0002-9394(02)02109-8. PMID12719097.
↑Michaelides, Michel; Holder, Graham E; Moore, Anthony T (2017), "Inherited retinal disorders", Taylor and Hoyt's Pediatric Ophthalmology and Strabismus, Elsevier, pp.462–486.e2, doi:10.1016/b978-0-7020-6616-0.00046-3, ISBN978-0-7020-6616-0
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.