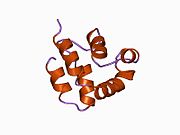
Caspases are a family of protease enzymes playing essential roles in programmed cell death. They are named caspases due to their specific cysteine protease activity – a cysteine in its active site nucleophilically attacks and cleaves a target protein only after an aspartic acid residue. As of 2009, there are 12 confirmed caspases in humans and 10 in mice, carrying out a variety of cellular functions.

Fas ligand is a type-II transmembrane protein expressed on various types of cells, including cytotoxic T lymphocytes, monocytes, neutrophils, breast epithelial cells, vascular endothelial cells and natural killer (NK) cells. It binds with its receptor, called FAS receptor and plays a crucial role in the regulation of the immune system and in induction of apoptosis, a programmed cell death.

The death-effector domain (DED) is a protein interaction domain found only in eukaryotes that regulates a variety of cellular signalling pathways. The DED domain is found in inactive procaspases and proteins that regulate caspase activation in the apoptosis cascade such as FAS-associating death domain-containing protein (FADD). FADD recruits procaspase 8 and procaspase 10 into a death induced signaling complex (DISC). This recruitment is mediated by a homotypic interaction between the procaspase DED and a second DED that is death effector domain in an adaptor protein that is directly associated with activated TNF receptors. Complex formation allows proteolytic activation of procaspase into the active caspase form which results in the initiation of apoptosis. Structurally the DED domain are a subclass of protein motif known as the death fold and contains 6 alpha helices, that closely resemble the structure of the Death domain (DD).

The death fold is a tertiary structure motif commonly found in proteins involved in apoptosis or inflammation-related processes. This motif is commonly found in domains that participate in protein–protein interactions leading to the formation of large functional complexes. Examples of death fold domains include the death domain (DD), death effector domain (DED), caspase recruitment domain (CARD), and pyrin domain (PYD).
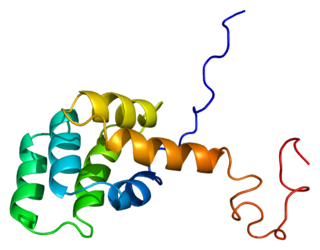
The Fas receptor, also known as Fas, FasR, apoptosis antigen 1, cluster of differentiation 95 (CD95) or tumor necrosis factor receptor superfamily member 6 (TNFRSF6), is a protein that in humans is encoded by the FAS gene. Fas was first identified using a monoclonal antibody generated by immunizing mice with the FS-7 cell line. Thus, the name Fas is derived from FS-7-associated surface antigen.

The death-inducing signaling complex (DISC) is a multi-protein complex formed by members of the death receptor family of apoptosis-inducing cellular receptors. A typical example is FasR, which forms the DISC upon trimerization as a result of its ligand (FasL) binding. The DISC is composed of the death receptor, FADD, and caspase 8. It transduces a downstream signal cascade resulting in apoptosis.

Caspase-8 is a caspase protein, encoded by the CASP8 gene. It most likely acts upon caspase-3. CASP8 orthologs have been identified in numerous mammals for which complete genome data are available. These unique orthologs are also present in birds.

Caspase-3 is a caspase protein that interacts with caspase-8 and caspase-9. It is encoded by the CASP3 gene. CASP3 orthologs have been identified in numerous mammals for which complete genome data are available. Unique orthologs are also present in birds, lizards, lissamphibians, and teleosts.
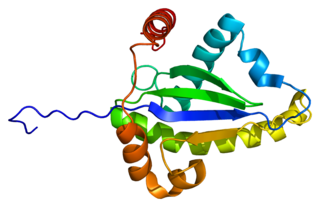
Tumor necrosis factor receptor type 1-associated DEATH domain protein is a protein that in humans is encoded by the TRADD gene.

Death receptor 4 (DR4), also known as TRAIL receptor 1 (TRAILR1) and tumor necrosis factor receptor superfamily member 10A (TNFRSF10A), is a cell surface receptor of the TNF-receptor superfamily that binds TRAIL and mediates apoptosis.

TNF receptor-associated factor 1 is a protein that in humans is encoded by the TRAF1 gene.

Caspase-10 is an enzyme that, in humans, is encoded by the CASP10 gene.

Death receptor 5 (DR5), also known as TRAIL receptor 2 (TRAILR2) and tumor necrosis factor receptor superfamily member 10B (TNFRSF10B), is a cell surface receptor of the TNF-receptor superfamily that binds TRAIL and mediates apoptosis.

Receptor-interacting serine/threonine-protein kinase 1 (RIPK1) functions in a variety of cellular pathways related to both cell survival and death. In terms of cell death, RIPK1 plays a role in apoptosis, necroptosis, and PANoptosis Some of the cell survival pathways RIPK1 participates in include NF-κB, Akt, and JNK.
Receptor-interacting serine/threonine-protein kinase 2 is an enzyme that in humans is encoded by the RIPK2 gene.

Astrocytic phosphoprotein PEA-15 is a protein that in humans is encoded by the PEA15 gene.

MAP kinase-activating death domain protein is an enzyme that in humans is encoded by the MADD gene. MADD is one out of four of the splice variants of the human IG20 (insulinoma-glucagonoma clone 20) gene which is located on human chromosome 11.

The death domain (DD) is a protein interaction module composed of a bundle of six alpha-helices. DD is a subclass of protein motif known as the death fold and is related in sequence and structure to the death effector domain (DED) and the caspase recruitment domain (CARD), which work in similar pathways and show similar interaction properties. DD bind each other forming oligomers. Mammals have numerous and diverse DD-containing proteins. Within these proteins, the DD domains can be found in combination with other domains, including: CARDs, DEDs, ankyrin repeats, caspase-like folds, kinase domains, leucine zippers, leucine-rich repeats (LRR), TIR domains, and ZU5 domains.
The Death Domain database is a secondary database of protein-protein interactions (PPI) of the death domain superfamily. Members of this superfamily are key players in apoptosis, inflammation, necrosis, and immune cell signaling pathways. Negative death domain superfamily-mediated signaling events result in various human diseases which include, cancers, neurodegenerative diseases, and immunological disorders. Creating death domain databases are of particular interest to researchers in the biomedical field as it enables a further understanding of the molecular mechanisms involved in death domain interactions while also providing easy access to tools such as an interaction map that illustrates the protein-protein interaction network and information. There is currently only one database that exclusively looks at death domains but there are other databases and resources that have information on this superfamily. According to PubMed, this database has been cited by seven peer-reviewed articles to date because of its extensive and specific information on the death domains and their PPI summaries.

AICD is programmed cell death caused by the interaction of Fas receptors and Fas ligands. AICD is a negative regulator of activated T lymphocytes that results from repeated stimulation of their T-cell receptors (TCR) and helps to maintain peripheral immune tolerance. Alteration of the process may lead to autoimmune diseases.