This article needs additional citations for verification .(January 2017) |
 | |
| Names | |
|---|---|
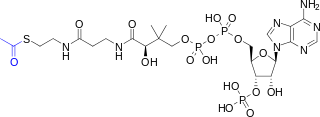
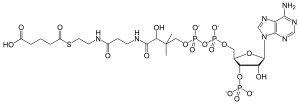
| IUPAC name 5-[(2-{3-[(2R)-4-{[1,3-Dihydroxy-1,3-dioxo-3-(3′-O-phosphonoadenosin-5′-O-yl)-1λ5,3λ5-diphosphoxan-1-yl]oxy}-3,3-dimethylbutanamido]propanamido}ethyl)sulfanyl]-5-oxopentanoic acid | |
| Systematic IUPAC name (9R)-1-[(2R,3S,4R,5R)-5-(6-Amino-9H-purin-9-yl)-4-hydroxy-3-(phosphonooxy)oxolan-2-yl]-3,5,9-trihydroxy-8,8-dimethyl-3,5,10,14,19-pentaoxo-2,4,6-trioxa-18-thia-11,15-diaza-3λ5,5λ5-diphosphatricosan-23-oic acid | |
| Identifiers | |
3D model (JSmol) | |
| ChemSpider | |
| MeSH | Glutaryl-coenzyme+A |
PubChem CID | |
CompTox Dashboard (EPA) | |
| |
| |
| Properties | |
| C26H42N7O19P3S | |
| Molar mass | 881.635 g/mol |
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
Glutaryl-coenzyme A is an intermediate in the metabolism of lysine and tryptophan. [1]