Related Research Articles

Coagulation, also known as clotting, is the process by which blood changes from a liquid to a gel, forming a blood clot. It potentially results in hemostasis, the cessation of blood loss from a damaged vessel, followed by repair. The mechanism of coagulation involves activation, adhesion and aggregation of platelets, as well as deposition and maturation of fibrin.
Factor V Leiden is a variant of human factor V, which causes an increase in blood clotting (hypercoagulability). Due to this mutation, protein C, an anticoagulant protein that normally inhibits the pro-clotting activity of factor V, is not able to bind normally to factor V, leading to a hypercoagulable state, i.e., an increased tendency for the patient to form abnormal and potentially harmful blood clots. Factor V Leiden is the most common hereditary hypercoagulability disorder amongst ethnic Europeans. It is named after the Dutch city of Leiden, where it was first identified in 1994 by Rogier Maria Bertina under the direction of Pieter Hendrick Reitsma. Despite the increased risk of venous thromboembolisms, people with one copy of this gene have not been found to have shorter lives than the general population. It is an autosomal dominant genetic disorder with incomplete penetrance.

Thrombin is a serine protease, an enzyme that, in humans, is encoded by the F2 gene. During the clotting process, prothrombin is proteolytically cleaved by the prothrombinase enzyme complex to form thrombin. Thrombin in turn acts as a serine protease that converts soluble fibrinogen into insoluble strands of fibrin, as well as catalyzing many other coagulation-related reactions.

Antithrombin (AT) is a small glycoprotein that inactivates several enzymes of the coagulation system. It is a 464-amino-acid protein produced by the liver. It contains three disulfide bonds and a total of four possible glycosylation sites. α-Antithrombin is the dominant form of antithrombin found in blood plasma and has an oligosaccharide occupying each of its four glycosylation sites. A single glycosylation site remains consistently un-occupied in the minor form of antithrombin, β-antithrombin. Its activity is increased manyfold by the anticoagulant drug heparin, which enhances the binding of antithrombin to factor IIa (thrombin) and factor Xa.
Low-molecular-weight heparin (LMWH) is a class of anticoagulant medications. They are used in the prevention of blood clots and treatment of venous thromboembolism and in the treatment of myocardial infarction.

Protein S is a vitamin K-dependent plasma glycoprotein synthesized in the liver. In the circulation, Protein S exists in two forms: a free form and a complex form bound to complement protein C4b-binding protein (C4BP). In humans, protein S is encoded by the PROS1 gene. Protein S plays a role in coagulation.
Protein C, also known as autoprothrombin IIA and blood coagulation factor XIV, is a zymogen, that is, an inactive enzyme. The activated form plays an important role in regulating anticoagulation, inflammation, and cell death and maintaining the permeability of blood vessel walls in humans and other animals. Activated protein C (APC) performs these operations primarily by proteolytically inactivating proteins Factor Va and Factor VIIIa. APC is classified as a serine protease since it contains a residue of serine in its active site. In humans, protein C is encoded by the PROC gene, which is found on chromosome 2.

Thrombophilia is an abnormality of blood coagulation that increases the risk of thrombosis. Such abnormalities can be identified in 50% of people who have an episode of thrombosis that was not provoked by other causes. A significant proportion of the population has a detectable thrombophilic abnormality, but most of these develop thrombosis only in the presence of an additional risk factor.

Factor X, also known by the eponym Stuart–Prower factor, is an enzyme of the coagulation cascade. It is a serine endopeptidase. Factor X is synthesized in the liver and requires vitamin K for its synthesis.
Factor V is a protein of the coagulation system, rarely referred to as proaccelerin or labile factor. In contrast to most other coagulation factors, it is not enzymatically active but functions as a cofactor. Deficiency leads to predisposition for hemorrhage, while some mutations predispose for thrombosis.

Hirudin is a naturally occurring peptide in the salivary glands of blood-sucking leeches that has a blood anticoagulant property. This is essential for the leeches' habit of feeding on blood, since it keeps a host's blood flowing after the worm's initial puncture of the skin.
Enteropeptidase is an enzyme produced by cells of the duodenum and is involved in digestion in humans and other animals. Enteropeptidase converts trypsinogen into its active form trypsin, resulting in the subsequent activation of pancreatic digestive enzymes. Absence of enteropeptidase results in intestinal digestion impairment.
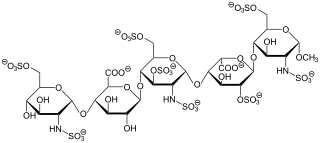
Fondaparinux is an anticoagulant medication chemically related to low molecular weight heparins. It is marketed by Viatris. A generic version developed by Alchemia is marketed within the US by Dr. Reddy's Laboratories.
Tissue factor pathway inhibitor is a single-chain polypeptide which can reversibly inhibit factor Xa (Xa). While Xa is inhibited, the Xa-TFPI complex can subsequently also inhibit the FVIIa-tissue factor complex. TFPI contributes significantly to the inhibition of Xa in vivo, despite being present at concentrations of only 2.5 nM.

Activated protein C resistance (APCR) is a hypercoagulability characterized by a lack of a response to activated protein C (APC), which normally helps prevent blood from clotting excessively. This results in an increased risk of venous thrombosis, which resulting in medical conditions such as deep vein thrombosis and pulmonary embolism. The most common cause of hereditary APC resistance is factor V Leiden mutation.

Heparin cofactor II (HCII), a protein encoded by the SERPIND1 gene, is a coagulation factor that inhibits IIa, and is a cofactor for heparin and dermatan sulfate.
Carboxypeptidase B2 (CPB2), also known as carboxypeptidase U (CPU), plasma carboxypeptidase B (pCPB) or thrombin-activatable fibrinolysis inhibitor (TAFI), is an enzyme that, in humans, is encoded by the gene CPB2.
Prothrombin fragment 1+2 (F1+2), also written as prothrombin fragment 1.2 (F1.2), is a polypeptide fragment of prothrombin generated by the in vivo cleavage of prothrombin into thrombin by the enzyme prothrombinase. It is released from the N-terminus of prothrombin. F1+2 is a marker of thrombin generation and hence of coagulation activation. It is considered the best marker of in vivo thrombin generation.
The activated protein C resistance (APCR) test is a coagulation test used in the evaluation and diagnosis of activated protein C (APC) resistance, a form of hypercoagulability. Hereditary APC resistance is usually caused by the factor V Leiden mutation, whereas acquired APC resistance has been linked to antiphospholipid antibodies, pregnancy, and estrogen therapy. APC resistance can be measured using either an activated partial thromboplastin time (aPTT)-based test or an endogenous thrombin potential (ETP)-based test.
Coagulation activation markers are biomarkers of net activation of coagulation and fibrinolysis. Examples include prothrombin fragment 1+2 (F1+2), thrombin–antithrombin complex (TAT), fibrinopeptide A (FpA), fibrin monomers (FMs), plasmin-α2-antiplasmin complex (PAP), activated protein C–protein C inhibitor (APC-PCI), and D-dimer (DD). These compounds are markers of thrombin generation, fibrin generation, and fibrinolysis. Coagulation activation markers, particularly D-dimer, are useful in the diagnosis of acute venous thromboembolism. They may also be useful in the assessment of hypercoagulability and venous thromboembolism risk.
References
- 1 2 Krishnaswamy S (January 2005). "Exosite-driven substrate specificity and function in coagulation". J. Thromb. Haemost. 3 (1): 54–67. doi: 10.1111/j.1538-7836.2004.01021.x . PMID 15634266. S2CID 478828.
- 1 2 Nesheim ME, Taswell JB, Mann KG (November 1979). "The contribution of bovine Factor V and Factor Va to the activity of prothrombinase". J. Biol. Chem. 254 (21): 10952–62. doi: 10.1016/S0021-9258(19)86616-4 . PMID 500617.
- ↑ Di Scipio RG, Kurachi K, Davie EW (June 1978). "Activation of human factor IX (Christmas factor)". J. Clin. Invest. 61 (6): 1528–38. doi:10.1172/JCI109073. PMC 372679 . PMID 659613.
- ↑ Furie B, Furie BC (May 1988). "The molecular basis of blood coagulation". Cell. 53 (4): 505–18. doi:10.1016/0092-8674(88)90567-3. PMID 3286010. S2CID 46652973.
- ↑ Di Scipio RG, Hermodson MA, Yates SG, Davie EW (February 1977). "A comparison of human prothrombin, factor IX (Christmas factor), factor X (Stuart factor), and protein S". Biochemistry. 16 (4): 698–706. doi:10.1021/bi00623a022. PMID 836809.
- ↑ Hoffman M, Monroe DM (June 2001). "A cell-based model of hemostasis". Thromb. Haemost. 85 (6): 958–65. doi:10.1055/s-0037-1615947. PMID 11434702. S2CID 18681597.
- ↑ Jenny RJ, Pittman DD, Toole JJ, Kriz RW, Aldape RA, Hewick RM, Kaufman RJ, Mann KG (July 1987). "Complete cDNA and derived amino acid sequence of human factor V". Proc. Natl. Acad. Sci. U.S.A. 84 (14): 4846–50. Bibcode:1987PNAS...84.4846J. doi: 10.1073/pnas.84.14.4846 . PMC 305202 . PMID 3110773.
- ↑ Mann KG, Kalafatis M (January 2003). "Factor V: a combination of Dr Jekyll and Mr Hyde". Blood. 101 (1): 20–30. doi: 10.1182/blood-2002-01-0290 . PMID 12393635.
- 1 2 3 Mann KG, Jenny RJ, Krishnaswamy S (1988). "Cofactor proteins in the assembly and expression of blood clotting enzyme complexes". Annu. Rev. Biochem. 57: 915–56. doi:10.1146/annurev.bi.57.070188.004411. PMID 3052293.
- 1 2 3 Krishnaswamy S (March 1990). "Prothrombinase complex assembly. Contributions of protein-protein and protein-membrane interactions toward complex formation". J. Biol. Chem. 265 (7): 3708–18. doi: 10.1016/S0021-9258(19)39652-8 . PMID 2303476.
- ↑ Majumder R, Quinn-Allen MA, Kane WH, Lentz BR (January 2005). "The phosphatidylserine binding site of the factor Va C2 domain accounts for membrane binding but does not contribute to the assembly or activity of a human factor Xa-factor Va complex". Biochemistry. 44 (2): 711–8. doi:10.1021/bi047962t. PMID 15641797.
- ↑ Autin L, Steen M, Dahlbäck B, Villoutreix BO (May 2006). "Proposed structural models of the prothrombinase (FXa-FVa) complex". Proteins. 63 (3): 440–50. doi:10.1002/prot.20848. PMID 16437549. S2CID 45651095.
- ↑ Krishnaswamy S, Mann KG, Nesheim ME (July 1986). "The prothrombinase-catalyzed activation of prothrombin proceeds through the intermediate meizothrombin in an ordered, sequential reaction". J. Biol. Chem. 261 (19): 8977–84. doi: 10.1016/S0021-9258(19)84477-0 . PMID 3755135.
- ↑ Rosing J, Tans G, Govers-Riemslag JW, Zwaal RF, Hemker HC (January 1980). "The role of phospholipids and factor Va in the prothrombinase complex". J. Biol. Chem. 255 (1): 274–83. doi: 10.1016/S0021-9258(19)86294-4 . PMID 7350159.
- ↑ van Rijn JL, Govers-Riemslag JW, Zwaal RF, Rosing J (September 1984). "Kinetic studies of prothrombin activation: effect of factor Va and phospholipids on the formation of the enzyme-substrate complex". Biochemistry. 23 (20): 4557–64. doi:10.1021/bi00315a008. PMID 6498156.
- ↑ Morita T, Iwanaga S (February 1978). "Purification and properties of prothrombin activator from the venom of Echis carinatus". J. Biochem. 83 (2): 559–70. doi:10.1093/oxfordjournals.jbchem.a131944. PMID 416016.
- 1 2 Esmon CT, Jackson CM (December 1974). "The conversion of prothrombin to thrombin. III. The factor Xa-catalyzed activation of prothrombin". J. Biol. Chem. 249 (24): 7782–90. doi: 10.1016/S0021-9258(19)42036-X . PMID 4430674.
- ↑ Krishnaswamy S, Church WR, Nesheim ME, Mann KG (March 1987). "Activation of human prothrombin by human prothrombinase. Influence of factor Va on the reaction mechanism". J. Biol. Chem. 262 (7): 3291–9. doi: 10.1016/S0021-9258(18)61503-0 . PMID 3818642.
- ↑ Guinto ER, Esmon CT (November 1984). "Loss of prothrombin and of factor Xa-factor Va interactions upon inactivation of factor Va by activated protein C". J. Biol. Chem. 259 (22): 13986–92. doi: 10.1016/S0021-9258(18)89842-8 . PMID 6438088.
- ↑ Krishnaswamy S, Williams EB, Mann KG (July 1986). "The binding of activated protein C to factors V and Va". J. Biol. Chem. 261 (21): 9684–93. doi: 10.1016/S0021-9258(18)67569-6 . PMID 3755431.
- ↑ Nesheim ME, Canfield WM, Kisiel W, Mann KG (February 1982). "Studies of the capacity of factor Xa to protect factor Va from inactivation by activated protein C". J. Biol. Chem. 257 (3): 1443–7. doi: 10.1016/S0021-9258(19)68213-X . PMID 6895752.
- ↑ van Wijk R, Nieuwenhuis K, van den Berg M, Huizinga EG, van der Meijden BB, Kraaijenhagen RJ, van Solinge WW (July 2001). "Five novel mutations in the gene for human blood coagulation factor V associated with type I factor V deficiency". Blood. 98 (2): 358–67. doi: 10.1182/blood.V98.2.358 . PMID 11435304. S2CID 1376514.
- ↑ Auerswald G (2006). "Prophylaxis in rare coagulation disorders -- factor X deficiency". Thromb. Res. 118 (Suppl 1): S29–31. doi:10.1016/j.thromres.2006.01.015. PMID 16574201.
- ↑ Kalafatis M, Rand MD, Mann KG (December 1994). "The mechanism of inactivation of human factor V and human factor Va by activated protein C". J. Biol. Chem. 269 (50): 31869–80. doi: 10.1016/S0021-9258(18)31776-9 . PMID 7989361.
- 1 2 3 Rosendorff A, Dorfman DM (June 2007). "Activated protein C resistance and factor V Leiden: a review". Arch. Pathol. Lab. Med. 131 (6): 866–71. doi:10.5858/2007-131-866-APCRAF. PMID 17550313.
- ↑ Mateo J, Oliver A, Borrell M, Sala N, Fontcuberta J (March 1997). "Laboratory evaluation and clinical characteristics of 2,132 consecutive unselected patients with venous thromboembolism--results of the Spanish Multicentric Study on Thrombophilia (EMET-Study)". Thromb. Haemost. 77 (3): 444–51. doi:10.1055/s-0038-1655986. PMID 9065991. S2CID 25695432.
- 1 2 Ornstein DL, Cushman M (April 2003). "Cardiology patient page. Factor V Leiden". Circulation. 107 (15): e94–7. doi: 10.1161/01.CIR.0000068167.08920.F1 . PMID 12707252.
- ↑ Folsom AR, Cushman M, Tsai MY, Aleksic N, Heckbert SR, Boland LL, Tsai AW, Yanez ND, Rosamond WD (April 2002). "A prospective study of venous thromboembolism in relation to factor V Leiden and related factors". Blood. 99 (8): 2720–5. doi: 10.1182/blood.V99.8.2720 . PMID 11929758. S2CID 12871240.
- ↑ Marchiori A, Mosena L, Prins MH, Prandoni P (August 2007). "The risk of recurrent venous thromboembolism among heterozygous carriers of factor V Leiden or prothrombin G20210A mutation. A systematic review of prospective studies". Haematologica. 92 (8): 1107–14. doi: 10.3324/haematol.10234 . PMID 17650440.
- ↑ Olson ST, Björk I, Sheffer R, Craig PA, Shore JD, Choay J (June 1992). "Role of the antithrombin-binding pentasaccharide in heparin acceleration of antithrombin-proteinase reactions. Resolution of the antithrombin conformational change contribution to heparin rate enhancement". J. Biol. Chem. 267 (18): 12528–38. doi: 10.1016/S0021-9258(18)42309-5 . PMID 1618758.
- ↑ Bauer KA (December 2003). "New pentasaccharides for prophylaxis of deep vein thrombosis: pharmacology". Chest. 124 (6 Suppl): 364S–370S. doi:10.1378/chest.124.6_suppl.364S. PMID 14668419.
- ↑ McRae SJ, Ginsberg JS (2005). "New Anticoagulants for the Prevention and Treatment of Venous Thromboembolism". Vasc Health Risk Manag. 1 (1): 41–53. doi: 10.2147/vhrm.1.1.41.58936 . PMC 1993925 . PMID 17319097.
- ↑ Kearon C (August 2004). "Long-term management of patients after venous thromboembolism". Circulation. 110 (9 Suppl 1): I10–8. doi: 10.1161/01.CIR.0000140902.46296.ae . PMID 15339876.
- ↑ Roehrig S, Straub A, Pohlmann J, Lampe T, Pernerstorfer J, Schlemmer KH, Reinemer P, Perzborn E (September 2005). "Discovery of the novel antithrombotic agent 5-chloro-N-({(5S)-2-oxo-3- [4-(3-oxomorpholin-4-yl)phenyl]-1,3-oxazolidin-5-yl}methyl)thiophene- 2-carboxamide (BAY 59-7939): an oral, direct factor Xa inhibitor". J. Med. Chem. 48 (19): 5900–8. doi:10.1021/jm050101d. PMID 16161994.
- 1 2 Pinto DJ, Orwat MJ, Koch S, Rossi KA, Alexander RS, Smallwood A, Wong PC, Rendina AR, Luettgen JM, Knabb RM, He K, Xin B, Wexler RR, Lam PY (November 2007). "Discovery of 1-(4-methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-1-yl)phenyl)-4,5,6,7-tetrahydro-1H-pyrazolo[3,4-c]pyridine-3-carboxamide (apixaban, BMS-562247), a highly potent, selective, efficacious, and orally bioavailable inhibitor of blood coagulation factor Xa". J. Med. Chem. 50 (22): 5339–56. doi:10.1021/jm070245n. PMID 17914785.
- ↑ Hauel NH, Nar H, Priepke H, Ries U, Stassen JM, Wienen W (April 2002). "Structure-based design of novel potent nonpeptide thrombin inhibitors". J. Med. Chem. 45 (9): 1757–66. doi:10.1021/jm0109513. PMID 11960487.
- ↑ Perzborn E, Strassburger J, Wilmen A, Pohlmann J, Roehrig S, Schlemmer KH, Straub A (March 2005). "In vitro and in vivo studies of the novel antithrombotic agent BAY 59-7939--an oral, direct Factor Xa inhibitor". J. Thromb. Haemost. 3 (3): 514–21. doi: 10.1111/j.1538-7836.2005.01166.x . PMID 15748242. S2CID 20809035.
- ↑ Gross PL, Weitz JI (March 2008). "New anticoagulants for treatment of venous thromboembolism". Arterioscler. Thromb. Vasc. Biol. 28 (3): 380–6. doi: 10.1161/ATVBAHA.108.162677 . PMID 18296593. S2CID 3074344.