Amenorrhea is the absence of a menstrual period in a female who has reached reproductive age. Physiological states of amenorrhoea are seen, most commonly, during pregnancy and lactation (breastfeeding). Outside the reproductive years, there is absence of menses during childhood and after menopause.
Luteinizing hormone is a hormone produced by gonadotropic cells in the anterior pituitary gland. The production of LH is regulated by gonadotropin-releasing hormone (GnRH) from the hypothalamus. In females, an acute rise of LH known as an LH surge, triggers ovulation and development of the corpus luteum. In males, where LH had also been called interstitial cell–stimulating hormone (ICSH), it stimulates Leydig cell production of testosterone. It acts synergistically with follicle-stimulating hormone (FSH).

Tuberous sclerosis complex (TSC) is a rare multisystem autosomal dominant genetic disease that causes non-cancerous tumours to grow in the brain and on other vital organs such as the kidneys, heart, liver, eyes, lungs and skin. A combination of symptoms may include seizures, intellectual disability, developmental delay, behavioral problems, skin abnormalities, lung disease, and kidney disease.
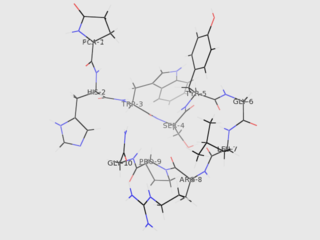
Gonadotropin-releasing hormone (GnRH) is a releasing hormone responsible for the release of follicle-stimulating hormone (FSH) and luteinizing hormone (LH) from the anterior pituitary. GnRH is a tropic peptide hormone synthesized and released from GnRH neurons within the hypothalamus. The peptide belongs to gonadotropin-releasing hormone family. It constitutes the initial step in the hypothalamic–pituitary–gonadal axis.
In medicine, precocious puberty is puberty occurring at an unusually early age. In most cases, the process is normal in every aspect except the unusually early age and simply represents a variation of normal development. In a minority of children with precocious puberty, the early development is triggered by a disease such as a tumor or injury of the brain. Even when there is no disease, unusually early puberty can have adverse effects on social behavior and psychological development, can reduce adult height potential, and may shift some lifelong health risks. Central precocious puberty can be treated by suppressing the pituitary hormones that induce sex steroid production. The opposite condition is delayed puberty.
Delayed puberty is when a person lacks or has incomplete development of specific sexual characteristics past the usual age of onset of puberty. The person may have no physical or hormonal signs that puberty has begun. In the United States, girls are considered to have delayed puberty if they lack breast development by age 13 or have not started menstruating by age 15. Boys are considered to have delayed puberty if they lack enlargement of the testicles by age 14. Delayed puberty affects about 2% of adolescents.

Hypopituitarism is the decreased (hypo) secretion of one or more of the eight hormones normally produced by the pituitary gland at the base of the brain. If there is decreased secretion of one specific pituitary hormone, the condition is known as selective hypopituitarism. If there is decreased secretion of most or all pituitary hormones, the term panhypopituitarism is used.
Gonadarche refers to the earliest gonadal changes of puberty. In response to pituitary gonadotropins, the ovaries in females and the testes in males begin to grow and increase the production of the sex steroids, especially estradiol and testosterone. The ovary and testis have receptors, follicle cells and leydig cells, respectively, where gonadotropins bind to stimulate the maturation of the gonads and secretion of estrogen and testosterone. Certain disorders can result in changes to timing or nature of these processes.

A hamartoma is a mostly benign, local malformation of cells that resembles a neoplasm of local tissue but is usually due to an overgrowth of multiple aberrant cells, with a basis in a systemic genetic condition, rather than a growth descended from a single mutated cell (monoclonality), as would typically define a benign neoplasm/tumor. Despite this, many hamartomas are found to have clonal chromosomal aberrations that are acquired through somatic mutations, and on this basis the term hamartoma is sometimes considered synonymous with neoplasm. Hamartomas are by definition benign, slow-growing or self-limiting, though the underlying condition may still predispose the individual towards malignancies.
Macroorchidism is a disorder found in males, specifically in children, where a subject has abnormally large testes. The condition is commonly inherited in connection with fragile X syndrome (FXS), which is also the second most common genetic cause of intellectual disability. The condition is also a rare sign of the McCune-Albright syndrome. The opposite of macroorchidism is called microorchidism, which is the condition of abnormally small testes.
Frontal lobe epilepsy (FLE) is a neurological disorder that is characterized by brief, recurring seizures arising in the frontal lobes of the brain, that often occur during sleep. It is the second most common type of epilepsy after temporal lobe epilepsy (TLE), and is related to the temporal form in that both forms are characterized by partial (focal) seizures.

Familial male-limited precocious puberty, often abbreviated as FMPP, also known as familial sexual precocity or gonadotropin-independent testotoxicosis, is a form of gonadotropin-independent precocious puberty in which boys experience early onset and progression of puberty. Signs of puberty can develop as early as an age of 1 year.

A pinealoma is a tumor of the pineal gland, a part of the brain that produces melatonin. If a pinealoma destroys the cells of the pineal gland in a child, it can cause precocious puberty.
Pallister–Hall syndrome (PHS) is a rare genetic disorder that affects various body systems. The main features are a non-cancerous mass on the hypothalamus and extra digits (polydactylism). The prevalence of Pallister-Hall Syndrome is unknown; about 100 cases have been reported in publication.
A gelastic seizure, also known as "gelastic epilepsy", is a rare type of seizure that involves a sudden burst of energy, usually in the form of laughing. This syndrome usually occurs for no obvious reason and is uncontrollable. It is slightly more common in males than females.
Hypothalamic disease is a disorder presenting primarily in the hypothalamus, which may be caused by damage resulting from malnutrition, including anorexia and bulimia eating disorders, genetic disorders, radiation, surgery, head trauma, lesion, tumour or other physical injury to the hypothalamus. The hypothalamus is the control center for several endocrine functions. Endocrine systems controlled by the hypothalamus are regulated by antidiuretic hormone (ADH), corticotropin-releasing hormone, gonadotropin-releasing hormone, growth hormone-releasing hormone, oxytocin, all of which are secreted by the hypothalamus. Damage to the hypothalamus may impact any of these hormones and the related endocrine systems. Many of these hypothalamic hormones act on the pituitary gland. Hypothalamic disease therefore affects the functioning of the pituitary and the target organs controlled by the pituitary, including the adrenal glands, ovaries and testes, and the thyroid gland.

Leydig cell hypoplasia (LCH), also known as Leydig cell agenesis, is a rare autosomal recessive genetic and endocrine syndrome affecting an estimated 1 in 1,000,000 genetic males. It is characterized by an inability of the body to respond to luteinizing hormone (LH), a gonadotropin which is normally responsible for signaling Leydig cells of the testicles to produce testosterone and other androgen sex hormones. The condition manifests itself as pseudohermaphroditism, hypergonadotropic hypogonadism, reduced or absent puberty, and infertility.
Gonadotropin-releasing hormone (GnRH) insensitivity also known as Isolated gonadotropin-releasing hormone (GnRH)deficiency (IGD) is a rare autosomal recessive genetic and endocrine syndrome which is characterized by inactivating mutations of the gonadotropin-releasing hormone receptor (GnRHR) and thus an insensitivity of the receptor to gonadotropin-releasing hormone (GnRH), resulting in a partial or complete loss of the ability of the gonads to synthesize the sex hormones. The condition manifests itself as isolated hypogonadotropic hypogonadism (IHH), presenting with symptoms such as delayed, reduced, or absent puberty, low or complete lack of libido, and infertility, and is the predominant cause of IHH when it does not present alongside anosmia.

Cavernous hemangioma, also called cavernous angioma, venous malformation, or cavernoma, is a type of venous malformation due to endothelial dysmorphogenesis from a lesion which is present at birth. A cavernoma in the brain is called a cerebral cavernous malformation or CCM. Despite its designation as a hemangioma, a cavernous hemangioma is not a tumor as it does not display endothelial hyperplasia. The abnormal tissue causes a slowing of blood flow through the cavities, or "caverns". The blood vessels do not form the necessary junctions with surrounding cells, and the structural support from the smooth muscle is hindered, causing leakage into the surrounding tissue. It is the leakage of blood, referred to as hemorrhage, that causes a variety of symptoms known to be associated with the condition.
Endoscopic endonasal surgery is a minimally invasive technique used mainly in neurosurgery and otolaryngology. A neurosurgeon or an otolaryngologist, using an endoscope that is entered through the nose, fixes or removes brain defects or tumors in the anterior skull base. Normally an otolaryngologist performs the initial stage of surgery through the nasal cavity and sphenoid bone; a neurosurgeon performs the rest of the surgery involving drilling into any cavities containing a neural organ such as the pituitary gland. The use of endoscope was first introduced in Transsphenoidal Pituitary Surgery by R Jankowsky, J Auque, C Simon et al. in 1992 G.



