
Proteomics is the large-scale study of proteins. Proteins are vital parts of living organisms, with many functions such as the formation of structural fibers of muscle tissue, enzymatic digestion of food, or synthesis and replication of DNA. In addition, other kinds of proteins include antibodies that protect an organism from infection, and hormones that send important signals throughout the body.

Tandem mass spectrometry, also known as MS/MS or MS2, is a technique in instrumental analysis where two or more stages of analysis using one or more mass analyzer are performed with an additional reaction step in between these analyses to increase their abilities to analyse chemical samples. A common use of tandem MS is the analysis of biomolecules, such as proteins and peptides.

Electron-capture dissociation (ECD) is a method of fragmenting gas-phase ions for structure elucidation of peptides and proteins in tandem mass spectrometry. It is one of the most widely used techniques for activation and dissociation of mass selected precursor ion in MS/MS. It involves the direct introduction of low-energy electrons to trapped gas-phase ions.

Citrullination or deimination is the conversion of the amino acid arginine in a protein into the amino acid citrulline. Citrulline is not one of the 20 standard amino acids encoded by DNA in the genetic code. Instead, it is the result of a post-translational modification. Citrullination is distinct from the formation of the free amino acid citrulline as part of the urea cycle or as a byproduct of enzymes of the nitric oxide synthase family.

In mass spectrometry, Orbitrap is an ion trap mass analyzer consisting of an outer barrel-like electrode and a coaxial inner spindle-like electrode that traps ions in an orbital motion around the spindle. The image current from the trapped ions is detected and converted to a mass spectrum using the Fourier transform of the frequency signal.
The Association of Biomolecular Resource Facilities (ABRF) is dedicated to advancing core and research biotechnology laboratories through research, communication, and education. ABRF members include over 2000 scientists representing 340 different core laboratories in 41 countries, including those in industry, government, academic and research institutions.

Rudolf Aebersold is a Swiss biologist, regarded as a pioneer in the fields of proteomics and systems biology. He has primarily researched techniques for measuring proteins in complex samples, in many cases via mass spectrometry. Ruedi Aebersold is a professor of Systems biology at the Institute of Molecular Systems Biology (IMSB) in ETH Zurich. He was one of the founders of the Institute for Systems Biology in Seattle, Washington, where he previously had a research group.

Electron-transfer dissociation (ETD) is a method of fragmenting multiply-charged gaseous macromolecules in a mass spectrometer between the stages of tandem mass spectrometry (MS/MS). Similar to electron-capture dissociation, ETD induces fragmentation of large, multiply-charged cations by transferring electrons to them. ETD is used extensively with polymers and biological molecules such as proteins and peptides for sequence analysis. Transferring an electron causes peptide backbone cleavage into c- and z-ions while leaving labile post translational modifications (PTM) intact. The technique only works well for higher charge state peptide or polymer ions (z>2). However, relative to collision-induced dissociation (CID), ETD is advantageous for the fragmentation of longer peptides or even entire proteins. This makes the technique important for top-down proteomics. The method was developed by Hunt and coworkers at the University of Virginia.
A tandem mass tag (TMT) is a chemical label that facilitates sample multiplexing in mass spectrometry (MS)-based quantification and identification of biological macromolecules such as proteins, peptides and nucleic acids. TMT belongs to a family of reagents referred to as isobaric mass tags which are a set of molecules with the same mass, but yield reporter ions of differing mass after fragmentation. The relative ratio of the measured reporter ions represents the relative abundance of the tagged molecule, although ion suppression has a detrimental effect on accuracy. Despite these complications, TMT-based proteomics has been shown to afford higher precision than Label-free quantification. In addition to aiding in protein quantification, TMT tags can also increase the detection sensitivity of certain highly hydrophilic analytes, such as phosphopeptides, in RPLC-MS analyses.

Protein mass spectrometry refers to the application of mass spectrometry to the study of proteins. Mass spectrometry is an important method for the accurate mass determination and characterization of proteins, and a variety of methods and instrumentations have been developed for its many uses. Its applications include the identification of proteins and their post-translational modifications, the elucidation of protein complexes, their subunits and functional interactions, as well as the global measurement of proteins in proteomics. It can also be used to localize proteins to the various organelles, and determine the interactions between different proteins as well as with membrane lipids.
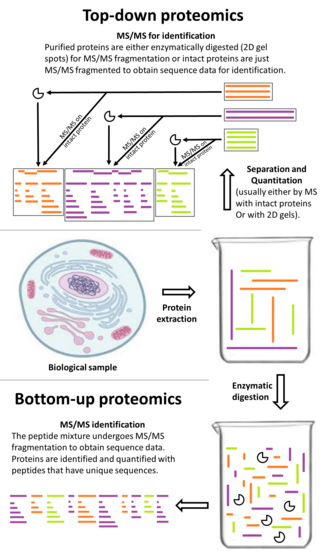
Top-down proteomics is a method of protein identification that either uses an ion trapping mass spectrometer to store an isolated protein ion for mass measurement and tandem mass spectrometry (MS/MS) analysis or other protein purification methods such as two-dimensional gel electrophoresis in conjunction with MS/MS. Top-down proteomics is capable of identifying and quantitating unique proteoforms through the analysis of intact proteins. The name is derived from the similar approach to DNA sequencing. During mass spectrometry intact proteins are typically ionized by electrospray ionization and trapped in a Fourier transform ion cyclotron resonance, quadrupole ion trap or Orbitrap mass spectrometer. Fragmentation for tandem mass spectrometry is accomplished by electron-capture dissociation or electron-transfer dissociation. Effective fractionation is critical for sample handling before mass-spectrometry-based proteomics. Proteome analysis routinely involves digesting intact proteins followed by inferred protein identification using mass spectrometry (MS). Top-down MS (non-gel) proteomics interrogates protein structure through measurement of an intact mass followed by direct ion dissociation in the gas phase.
Bottom-up proteomics is a common method to identify proteins and characterize their amino acid sequences and post-translational modifications by proteolytic digestion of proteins prior to analysis by mass spectrometry. The major alternative workflow used in proteomics is called top-down proteomics where intact proteins are purified prior to digestion and/or fragmentation either within the mass spectrometer or by 2D electrophoresis. Essentially, bottom-up proteomics is a relatively simple and reliable means of determining the protein make-up of a given sample of cells, tissues, etc.

Laser spray ionization refers to one of several methods for creating ions using a laser interacting with a spray of neutral particles or ablating material to create a plume of charged particles. The ions thus formed can be separated by m/z with mass spectrometry. Laser spray is one of several ion sources that can be coupled with liquid chromatography-mass spectrometry for the detection of larger molecules.

Selected reaction monitoring (SRM), also called multiple reaction monitoring (MRM), is a method used in tandem mass spectrometry in which an ion of a particular mass is selected in the first stage of a tandem mass spectrometer and an ion product of a fragmentation reaction of the precursor ions is selected in the second mass spectrometer stage for detection.

In the field of cellular biology, single-cell analysis and subcellular analysis is the study of genomics, transcriptomics, proteomics, metabolomics and cell–cell interactions at the single cell level. The concept of single-cell analysis originated in the 1970s. Before the discovery of heterogeneity, single-cell analysis mainly referred to the analysis or manipulation of an individual cell in a bulk population of cells at a particular condition using optical or electronic microscope. To date, due to the heterogeneity seen in both eukaryotic and prokaryotic cell populations, analyzing a single cell makes it possible to discover mechanisms not seen when studying a bulk population of cells. Technologies such as fluorescence-activated cell sorting (FACS) allow the precise isolation of selected single cells from complex samples, while high throughput single cell partitioning technologies, enable the simultaneous molecular analysis of hundreds or thousands of single unsorted cells; this is particularly useful for the analysis of transcriptome variation in genotypically identical cells, allowing the definition of otherwise undetectable cell subtypes. The development of new technologies is increasing our ability to analyze the genome and transcriptome of single cells, as well as to quantify their proteome and metabolome. Mass spectrometry techniques have become important analytical tools for proteomic and metabolomic analysis of single cells. Recent advances have enabled quantifying thousands of protein across hundreds of single cells, and thus make possible new types of analysis. In situ sequencing and fluorescence in situ hybridization (FISH) do not require that cells be isolated and are increasingly being used for analysis of tissues.
The Bijvoet Centre for Biomolecular Research is a research institute at Utrecht University. The Bijvoet Centre performs research on the relation between the structure and function of biomolecules, including proteins and lipids, which play a role in biological processes such as regulation, interaction and recognition. The Bijvoet Centre houses advanced infrastructures for the analysis of proteins and other biomolecules using NMR, X-ray crystallography, electron microscopy and mass spectrometry. The institute is named after famous Dutch chemist Johannes Martin Bijvoet, who worked at Utrecht University.
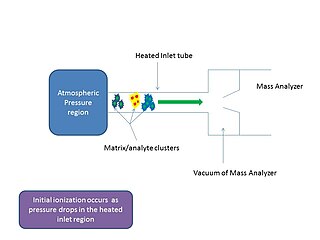
In mass spectrometry, matrix-assisted ionization is a low fragmentation (soft) ionization technique which involves the transfer of particles of the analyte and matrix sample from atmospheric pressure (AP) to the heated inlet tube connecting the AP region to the vacuum of the mass analyzer.

Ancient proteins are complex mixtures and the term palaeoproteomics is used to characterise the study of proteomes in the past. Ancients proteins have been recovered from a wide range of archaeological materials, including bones, teeth, eggshells, leathers, parchments, ceramics, painting binders and well-preserved soft tissues like gut intestines. These preserved proteins have provided valuable information about taxonomic identification, evolution history (phylogeny), diet, health, disease, technology and social dynamics in the past.

Ron M.A. Heeren is a Dutch scientist in mass spectrometry imaging. He is currently a distinguished professor at Maastricht University and the scientific director of the Multimodal Molecular Imaging Institute (M4I), where he heads the division of Imaging Mass Spectrometry.
