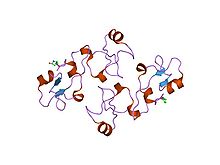
An alpha helix is a sequence of amino acids in a protein that are twisted into a coil.
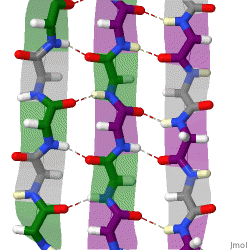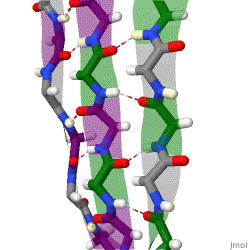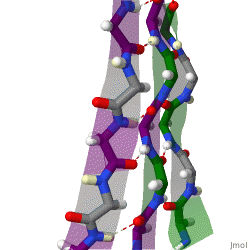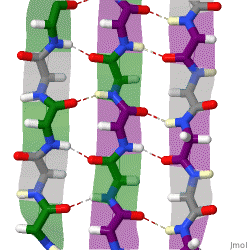
The beta sheet, (β-sheet) is a common motif of the regular protein secondary structure. Beta sheets consist of beta strands (β-strands) connected laterally by at least two or three backbone hydrogen bonds, forming a generally twisted, pleated sheet. A β-strand is a stretch of polypeptide chain typically 3 to 10 amino acids long with backbone in an extended conformation. The supramolecular association of β-sheets has been implicated in the formation of the fibrils and protein aggregates observed in amyloidosis, Alzheimer's disease and other proteinopathies.

Proteins are large biomolecules and macromolecules that comprise one or more long chains of amino acid residues. Proteins perform a vast array of functions within organisms, including catalysing metabolic reactions, DNA replication, responding to stimuli, providing structure to cells and organisms, and transporting molecules from one location to another. Proteins differ from one another primarily in their sequence of amino acids, which is dictated by the nucleotide sequence of their genes, and which usually results in protein folding into a specific 3D structure that determines its activity.

Protein tertiary structure is the three dimensional shape of a protein. The tertiary structure will have a single polypeptide chain "backbone" with one or more protein secondary structures, the protein domains. Amino acid side chains may interact and bond in a number of ways. The interactions and bonds of side chains within a particular protein determine its tertiary structure. The protein tertiary structure is defined by its atomic coordinates. These coordinates may refer either to a protein domain or to the entire tertiary structure. A number of tertiary structures may fold into a quaternary structure.
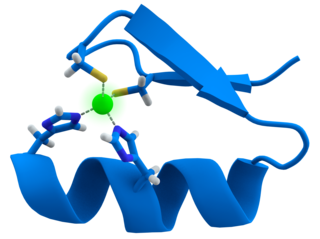
A zinc finger is a small protein structural motif that is characterized by the coordination of one or more zinc ions (Zn2+) which stabilizes the fold. It was originally coined to describe the finger-like appearance of a hypothesized structure from the African clawed frog (Xenopus laevis) transcription factor IIIA. However, it has been found to encompass a wide variety of differing protein structures in eukaryotic cells. Xenopus laevis TFIIIA was originally demonstrated to contain zinc and require the metal for function in 1983, the first such reported zinc requirement for a gene regulatory protein followed soon thereafter by the Krüppel factor in Drosophila. It often appears as a metal-binding domain in multi-domain proteins.

Protein structure prediction is the inference of the three-dimensional structure of a protein from its amino acid sequence—that is, the prediction of its secondary and tertiary structure from primary structure. Structure prediction is different from the inverse problem of protein design. Protein structure prediction is one of the most important goals pursued by computational biology; and it is important in medicine and biotechnology.
In biology and biochemistry, protease inhibitors, or antiproteases, are molecules that inhibit the function of proteases. Many naturally occurring protease inhibitors are proteins.
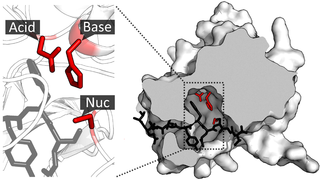
A catalytic triad is a set of three coordinated amino acids that can be found in the active site of some enzymes. Catalytic triads are most commonly found in hydrolase and transferase enzymes. An acid-base-nucleophile triad is a common motif for generating a nucleophilic residue for covalent catalysis. The residues form a charge-relay network to polarise and activate the nucleophile, which attacks the substrate, forming a covalent intermediate which is then hydrolysed to release the product and regenerate free enzyme. The nucleophile is most commonly a serine or cysteine amino acid, but occasionally threonine or even selenocysteine. The 3D structure of the enzyme brings together the triad residues in a precise orientation, even though they may be far apart in the sequence.
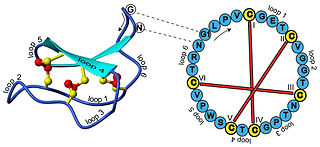
In biochemistry, cyclotides are small, disulfide-rich peptides isolated from plants. Typically containing 28-37 amino acids, they are characterized by their head-to-tail cyclised peptide backbone and the interlocking arrangement of their three disulfide bonds. These combined features have been termed the cyclic cystine knot (CCK) motif. To date, over 100 cyclotides have been isolated and characterized from species of the families Rubiaceae, Violaceae, and Cucurbitaceae. Cyclotides have also been identified in agriculturally important families such as the Fabaceae and Poaceae.
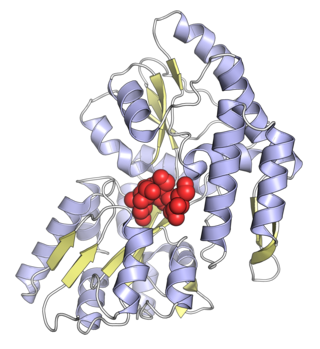
Maltose-binding protein (MBP) is a part of the maltose/maltodextrin system of Escherichia coli, which is responsible for the uptake and efficient catabolism of maltodextrins. It is a complex regulatory and transport system involving many proteins and protein complexes. MBP has an approximate molecular mass of 42.5 kilodaltons.

In molecular biology, a protein domain is a region of a protein's polypeptide chain that is self-stabilizing and that folds independently from the rest. Each domain forms a compact folded three-dimensional structure. Many proteins consist of several domains, and a domain may appear in a variety of different proteins. Molecular evolution uses domains as building blocks and these may be recombined in different arrangements to create proteins with different functions. In general, domains vary in length from between about 50 amino acids up to 250 amino acids in length. The shortest domains, such as zinc fingers, are stabilized by metal ions or disulfide bridges. Domains often form functional units, such as the calcium-binding EF hand domain of calmodulin. Because they are independently stable, domains can be "swapped" by genetic engineering between one protein and another to make chimeric proteins.

In enzymology, a protein-glutamate O-methyltransferase is an enzyme that catalyzes the chemical reaction

Trefoil factor 1 is a protein that in humans is encoded by the TFF1 gene.

Trefoil factor 2 is a protein that in humans is encoded by the TFF2 gene.

A circular permutation is a relationship between proteins whereby the proteins have a changed order of amino acids in their peptide sequence. The result is a protein structure with different connectivity, but overall similar three-dimensional (3D) shape. In 1979, the first pair of circularly permuted proteins – concanavalin A and lectin – were discovered; over 2000 such proteins are now known.

The SET domain is a protein domain that typically has methyltransferase activity. It was originally identified as part of a larger conserved region present in the Drosophila Trithorax protein and was subsequently identified in the Drosophila Su(var)3-9 and 'Enhancer of zeste' proteins, from which the acronym SET is derived [Su(var)3-9, Enhancer-of-zeste and Trithorax].
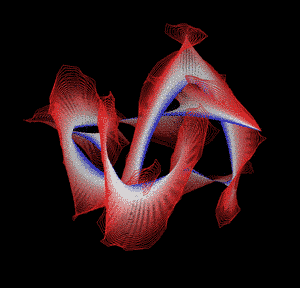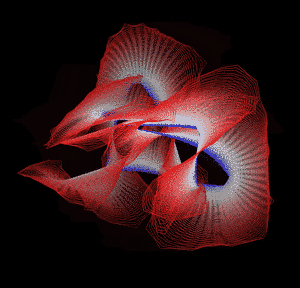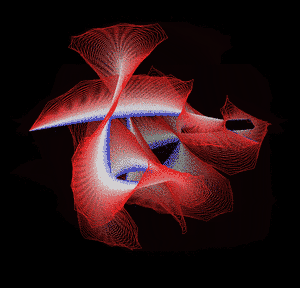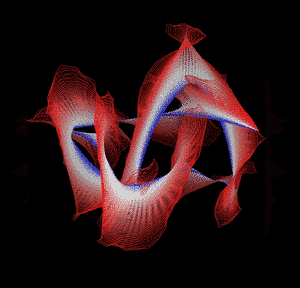
Knotted proteins are proteins whose backbones entangle themselves in a knot. One can imagine pulling a protein chain from both termini, as though pulling a string from both ends. When a knotted protein is “pulled” from both termini, it does not get disentangled. Knotted proteins are very rare, making up only about one percent of the proteins in the Protein Data Bank, and their folding mechanisms and function are not well understood. Although there are experimental and theoretical studies that hint to some answers, systematic answers to these questions have not yet been found.
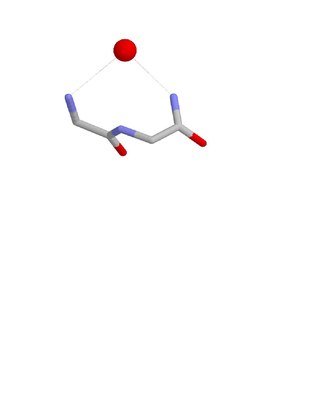
The Nest is a type of protein structural motif. It is a small recurring anion-binding feature of both proteins and peptides. Each consists of the main chain atoms of three consecutive amino acid residues. The main chain NH groups bind the anions while the side chain atoms are often not involved. Proline residues lack NH groups so are rare in nests. About one in 12 of amino acid residues in proteins, on average, belongs to a nest.
Atrolysin A is an enzyme that is one of six hemorrhagic toxins found in the venom of western diamondback rattlesnake. This endopeptidase has a length of 419 amino acid residues. The metalloproteinase disintegrin-like domain and the cysteine-rich domain of the enzyme are responsible for the enzyme's hemorrhagic effects on organisms via inhibition of platelet aggregation.
A protein superfamily is the largest grouping (clade) of proteins for which common ancestry can be inferred. Usually this common ancestry is inferred from structural alignment and mechanistic similarity, even if no sequence similarity is evident. Sequence homology can then be deduced even if not apparent. Superfamilies typically contain several protein families which show sequence similarity within each family. The term protein clan is commonly used for protease and glycosyl hydrolases superfamilies based on the MEROPS and CAZy classification systems.