In biology, histones are highly basic proteins abundant in lysine and arginine residues that are found in eukaryotic cell nuclei and in most Archaeal phyla. They act as spools around which DNA winds to create structural units called nucleosomes. Nucleosomes in turn are wrapped into 30-nanometer fibers that form tightly packed chromatin. Histones prevent DNA from becoming tangled and protect it from DNA damage. In addition, histones play important roles in gene regulation and DNA replication. Without histones, unwound DNA in chromosomes would be very long. For example, each human cell has about 1.8 meters of DNA if completely stretched out; however, when wound about histones, this length is reduced to about 90 micrometers (0.09 mm) of 30 nm diameter chromatin fibers.
Methylation, in the chemical sciences, is the addition of a methyl group on a substrate, or the substitution of an atom by a methyl group. Methylation is a form of alkylation, with a methyl group replacing a hydrogen atom. These terms are commonly used in chemistry, biochemistry, soil science, and biology.
In biochemistry, the DNA methyltransferase family of enzymes catalyze the transfer of a methyl group to DNA. DNA methylation serves a wide variety of biological functions. All the known DNA methyltransferases use S-adenosyl methionine (SAM) as the methyl donor.

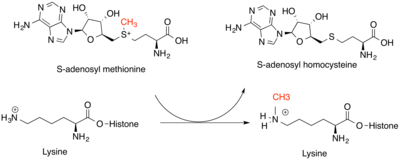
Histone methyltransferases (HMT) are histone-modifying enzymes, that catalyze the transfer of one, two, or three methyl groups to lysine and arginine residues of histone proteins. The attachment of methyl groups occurs predominantly at specific lysine or arginine residues on histones H3 and H4. Two major types of histone methyltranferases exist, lysine-specific and arginine-specific. In both types of histone methyltransferases, S-Adenosyl methionine (SAM) serves as a cofactor and methyl donor group.
The genomic DNA of eukaryotes associates with histones to form chromatin. The level of chromatin compaction depends heavily on histone methylation and other post-translational modifications of histones. Histone methylation is a principal epigenetic modification of chromatin that determines gene expression, genomic stability, stem cell maturation, cell lineage development, genetic imprinting, DNA methylation, and cell mitosis.
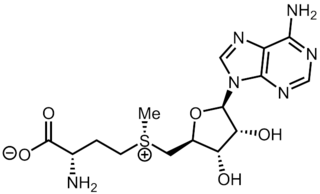
S-Adenosyl methionine (SAM), also known under the commercial names of SAMe, SAM-e, or AdoMet, is a common cosubstrate involved in methyl group transfers, transsulfuration, and aminopropylation. Although these anabolic reactions occur throughout the body, most SAM is produced and consumed in the liver. More than 40 methyl transfers from SAM are known, to various substrates such as nucleic acids, proteins, lipids and secondary metabolites. It is made from adenosine triphosphate (ATP) and methionine by methionine adenosyltransferase. SAM was first discovered by Giulio Cantoni in 1952.

DNA methylation is a biological process by which methyl groups are added to the DNA molecule. Methylation can change the activity of a DNA segment without changing the sequence. When located in a gene promoter, DNA methylation typically acts to repress gene transcription. In mammals, DNA methylation is essential for normal development and is associated with a number of key processes including genomic imprinting, X-chromosome inactivation, repression of transposable elements, aging, and carcinogenesis.

In biology, the epigenome of an organism is the collection of chemical changes to its DNA and histone proteins that affects when, where, and how the DNA is expressed; these changes can be passed down to an organism's offspring via transgenerational epigenetic inheritance. Changes to the epigenome can result in changes to the structure of chromatin and changes to the function of the genome. The human epigenome, including DNA methylation and histone modification, is maintained through cell division. The epigenome is essential for normal development and cellular differentiation, enabling cells with the same genetic code to perform different functions. The human epigenome is dynamic and can be influenced by environmental factors such as diet, stress, and toxins.
Histone methylation is a process by which methyl groups are transferred to amino acids of histone proteins that make up nucleosomes, which the DNA double helix wraps around to form chromosomes. Methylation of histones can either increase or decrease transcription of genes, depending on which amino acids in the histones are methylated, and how many methyl groups are attached. Methylation events that weaken chemical attractions between histone tails and DNA increase transcription because they enable the DNA to uncoil from nucleosomes so that transcription factor proteins and RNA polymerase can access the DNA. This process is critical for the regulation of gene expression that allows different cells to express different genes.

DNA adenine methylase, (Dam) (also site-specific DNA-methyltransferase (adenine-specific), EC 2.1.1.72, modification methylase, restriction-modification system) is an enzyme that adds a methyl group to the adenine of the sequence 5'-GATC-3' in newly synthesized DNA. Immediately after DNA synthesis, the daughter strand remains unmethylated for a short time. It is an orphan methyltransferase that is not part of a restriction-modification system and regulates gene expression. This enzyme catalyses the following chemical reaction

In enzymology, a protein-glutamate O-methyltransferase is an enzyme that catalyzes the chemical reaction
In enzymology, a tRNA (cytosine-5-)-methyltransferase is an enzyme that catalyzes the chemical reaction

Enhancer of zeste homolog 2 (EZH2) is a histone-lysine N-methyltransferase enzyme encoded by EZH2 gene, that participates in histone methylation and, ultimately, transcriptional repression. EZH2 catalyzes the addition of methyl groups to histone H3 at lysine 27, by using the cofactor S-adenosyl-L-methionine. Methylation activity of EZH2 facilitates heterochromatin formation thereby silences gene function. Remodeling of chromosomal heterochromatin by EZH2 is also required during cell mitosis.

In molecular biology, a Tudor domain is a conserved protein structural domain originally identified in the Tudor protein encoded in Drosophila. The Tudor gene was found in a Drosophila screen for maternal factors that regulate embryonic development or fertility. Mutations here are lethal for offspring, inspiring the name Tudor, as a reference to the Tudor King Henry VIII and the several miscarriages experienced by his wives.

The SET domain is a protein domain that typically has methyltransferase activity. It was originally identified as part of a larger conserved region present in the Drosophila Trithorax protein and was subsequently identified in the Drosophila Su(var)3-9 and 'Enhancer of zeste' proteins, from which the acronym SET is derived [Su(var)3-9, Enhancer-of-zeste and Trithorax].

Euchromatic histone-lysine N-methyltransferase 1, also known as G9a-like protein (GLP), is a protein that in humans is encoded by the EHMT1 gene.
While the cellular and molecular mechanisms of learning and memory have long been a central focus of neuroscience, it is only in recent years that attention has turned to the epigenetic mechanisms behind the dynamic changes in gene transcription responsible for memory formation and maintenance. Epigenetic gene regulation often involves the physical marking of DNA or associated proteins to cause or allow long-lasting changes in gene activity. Epigenetic mechanisms such as DNA methylation and histone modifications have been shown to play an important role in learning and memory.
Protein methylation is a type of post-translational modification featuring the addition of methyl groups to proteins. It can occur on the nitrogen-containing side-chains of arginine and lysine, but also at the amino- and carboxy-termini of a number of different proteins. In biology, methyltransferases catalyze the methylation process, activated primarily by S-adenosylmethionine. Protein methylation has been most studied in histones, where the transfer of methyl groups from S-adenosyl methionine is catalyzed by histone methyltransferases. Histones that are methylated on certain residues can act epigenetically to repress or activate gene expression.
H3K4me3 is an epigenetic modification to the DNA packaging protein Histone H3 that indicates tri-methylation at the 4th lysine residue of the histone H3 protein and is often involved in the regulation of gene expression. The name denotes the addition of three methyl groups (trimethylation) to the lysine 4 on the histone H3 protein.

Thomas Jenuwein is a German scientist working in the fields of epigenetics, chromatin biology, gene regulation and genome function.
Transgenerational epigenetic inheritance in plants involves mechanisms for the passing of epigenetic marks from parent to offspring that differ from those reported in animals. There are several kinds of epigenetic markers, but they all provide a mechanism to facilitate greater phenotypic plasticity by influencing the expression of genes without altering the DNA code. These modifications represent responses to environmental input and are reversible changes to gene expression patterns that can be passed down through generations. In plants, transgenerational epigenetic inheritance could potentially represent an evolutionary adaptation for sessile organisms to quickly adapt to their changing environment.