
The coenzyme Q : cytochrome c – oxidoreductase, sometimes called the cytochrome bc1 complex, and at other times complex III, is the third complex in the electron transport chain, playing a critical role in biochemical generation of ATP. Complex III is a multisubunit transmembrane protein encoded by both the mitochondrial and the nuclear genomes. Complex III is present in the mitochondria of all animals and all aerobic eukaryotes and the inner membranes of most eubacteria. Mutations in Complex III cause exercise intolerance as well as multisystem disorders. The bc1 complex contains 11 subunits, 3 respiratory subunits, 2 core proteins and 6 low-molecular weight proteins.

Cytochrome c oxidase I (COX1) also known as mitochondrially encoded cytochrome c oxidase I (MT-CO1) is a protein that is encoded by the MT-CO1 gene in eukaryotes. The gene is also called COX1, CO1, or COI. Cytochrome c oxidase I is the main subunit of the cytochrome c oxidase complex. In humans, mutations in MT-CO1 have been associated with Leber's hereditary optic neuropathy (LHON), acquired idiopathic sideroblastic anemia, Complex IV deficiency, colorectal cancer, sensorineural deafness, and recurrent myoglobinuria.

Cytochrome c oxidase subunit III (COX3) is an enzyme that in humans is encoded by the MT-CO3 gene. It is one of main transmembrane subunits of cytochrome c oxidase. It is also one of the three mitochondrial DNA (mtDNA) encoded subunits of respiratory complex IV. Variants of it have been associated with isolated myopathy, severe encephalomyopathy, Leber hereditary optic neuropathy, mitochondrial complex IV deficiency, and recurrent myoglobinuria.
Cytochrome b is a protein that in humans is encoded by the MT-CYB gene. Its gene product is a subunit of the respiratory chain protein ubiquinol–cytochrome c reductase, which consists of the products of one mitochondrially encoded gene, MT-CYB, and ten nuclear genes—UQCRC1, UQCRC2, CYC1, UQCRFS1, UQCRB, "11kDa protein", UQCRH, Rieske protein presequence, "cyt c1 associated protein", and Rieske-associated protein.
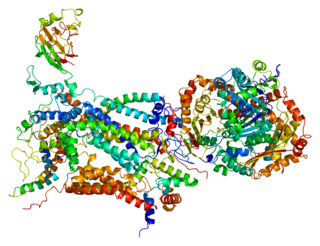
Cytochrome c1, heme protein, mitochondrial (CYC1), also known as UQCR4, MC3DN6, Complex III subunit 4, Cytochrome b-c1 complex subunit 4, or Ubiquinol-cytochrome-c reductase complex cytochrome c1 subunit is a protein that in humans is encoded by the CYC1 gene. CYC1 is a respiratory subunit of Ubiquinol Cytochrome c Reductase, which is located in the inner mitochondrial membrane and is part of the electron transport chain. Mutations in this gene may cause mitochondrial complex III deficiency, nuclear, type 6.

NADH dehydrogenase [ubiquinone] iron-sulfur protein 4, mitochondrial (NDUFS4) also known as NADH-ubiquinone oxidoreductase 18 kDa subunit is an enzyme that in humans is encoded by the NDUFS4 gene. This gene encodes a nuclear-encoded accessory subunit of the mitochondrial membrane respiratory chain NADH dehydrogenase. Complex I removes electrons from NADH and passes them to the electron acceptor ubiquinone. Mutations in this gene can cause mitochondrial complex I deficiencies such as Leigh syndrome.

Surfeit locus protein 1 (SURF1) is a protein that in humans is encoded by the SURF1 gene. The protein encoded by SURF1 is a component of the mitochondrial translation regulation assembly intermediate of cytochrome c oxidase complex, which is involved in the regulation of cytochrome c oxidase assembly. Defects in this gene are a cause of Leigh syndrome, a severe neurological disorder that is commonly associated with systemic cytochrome c oxidase deficiency, and Charcot-Marie-Tooth disease 4K (CMT4K).

Cytochrome b-c1 complex subunit 1, mitochondrial is a protein that in humans is encoded by the UQCRC1 gene.

Ubiquinol-cytochrome c reductase binding protein, also known as UQCRB, Complex III subunit 7, QP-C, or Ubiquinol-cytochrome c reductase complex 14 kDa protein is a protein which in humans is encoded by the UQCRB gene. This gene encodes a subunit of the ubiquinol-cytochrome c oxidoreductase complex, which consists of one mitochondrial-encoded and 10 nuclear-encoded subunits. Mutations in this gene are associated with mitochondrial complex III deficiency. Alternatively spliced transcript variants have been found for this gene. Related pseudogenes have been identified on chromosomes 1, 5 and X.

Cytochrome b-c1 complex subunit 2, mitochondrial (UQCRC2), also known as QCR2, UQCR2, or MC3DN5 is a protein that in humans is encoded by the UQCRC2 gene. The product of UQCRC2 is a subunit of the respiratory chain protein Ubiquinol Cytochrome c Reductase, which consists of the products of one mitochondrially encoded gene, MTCYTB and ten nuclear genes: UQCRC1, UQCRC2, Cytochrome c1, UQCRFS1, UQCRB, "11kDa protein", UQCRH, Rieske Protein presequence, "cyt. c1 associated protein", and "Rieske-associated protein." Defects in UQCRC2 are associated with mitochondrial complex III deficiency, nuclear, type 5.

Ubiquinol-cytochrome c reductase, Rieske iron-sulfur polypeptide 1, also known as UQCRFS1, Rieske iron-sulfur (Fe-S) protein, Cytochrome b-c1 complex subunit 5, or Complex III subunit 5 is a protein which in humans is encoded by the UQCRFS1 gene. UQCRFS1 is a subunit of the respiratory chain protein Ubiquinol Cytochrome c Reductase, which consists of the products of one mitochondrially encoded gene, MTCYTB and ten nuclear genes UQCRC1, UQCRC2, Cytochrome C1, UQCRFS1, UQCRB,UQCRQ, UQCRH, UCRC, and UQCR.

Cytochrome c oxidase assembly protein COX15 homolog (COX15), also known as heme A synthase, is a protein that in humans is encoded by the COX15 gene. This protein localizes to the inner mitochondrial membrane and involved in heme A biosynthesis. COX15 is also part of a three-component mono-oxygenase that catalyses the hydroxylation of the methyl group at position eight of the protoheme molecule. Mutations in this gene has been reported in patients with hypertrophic cardiomyopathy as well as Leigh syndrome, and characterized by delayed onset of symptoms, hypotonia, feeding difficulties, failure to thrive, motor regression, and brain stem signs.
Björnstad syndrome is an autosomal recessive congenital condition involving pili torti, sensorineural deafness, and hair abnormalities. It was first characterized in 1965, in Oslo, by prof. Roar Theodor Bjørnstad after he observed an association between pili torti and hearing loss. The condition is extremely rare, with less than 50 cases documented in medical literature worldwide.
Ubiquinol-cytochrome c reductase, complex III subunit VII, 9.5kDa is a protein that in humans is encoded by the UQCRQ gene. This ubiqinone-binding protein is a subunit of mitochondrial Complex III in the electron transport chain. A mutation in the UQCRQ gene has been shown to cause severe neurological disorders. Infection by Trypanosoma cruzi can cause oxidative modification of this protein in cardiac muscle tissue.

Succinate dehydrogenase complex assembly factor 1 (SDHAF1), also known as LYR motif-containing protein 8 (LYRM8), is a protein that in humans is encoded by the SDHAF1, or LYRM8, gene. SDHAF1 is a chaperone protein involved in the assembly of the succinate dehydrogenase (SDH) complex. Mutations in this gene are associated with SDH-defective infantile leukoencephalopathy and mitochondrial complex II deficiency.

Tetratricopeptide repeat domain 19, also known as TPR repeat protein 19 or Tetratricopeptide repeat protein 19, mitochondrial is a protein that in humans is encoded by the TTC19 gene. This gene encodes a protein with a tetratricopeptide repeat (TPR) domain containing several TPRs of about 34 amino acids each. These repeats are found in a variety of organisms including bacteria, fungi and plants, and are involved in a variety of functions including protein-protein interactions. This protein is embedded in the inner mitochondrial membrane and is involved in the formation of the mitochondrial respiratory chain III. It has also been suggested that this protein plays a role in cytokinesis. Mutations in this gene cause mitochondrial complex III deficiency. Alternatively spliced transcript variants have been found for this gene.

Ubiquinol-cytochrome c reductase complex assembly factor 2 is a protein that in humans is encoded by the UQCC2 gene. Located in the mitochondrial nucleoid, this protein is a complex III assembly factor, playing a role in cytochrome b biogenesis along with the UQCC1 protein. It regulates insulin secretion and mitochondrial ATP production and oxygen consumption. In the sole recorded case, a mutation in the UQCC2 gene caused Complex III deficiency, characterized by intrauterine growth retardation, neonatal lactic acidosis, and renal tubular dysfunction.
LYR motif containing 7, also known as Complex III assembly factor LYRM7 or LYR motif-containing protein 7 is a protein that in humans is encoded by the LYRM7 gene. The protein encoded by this gene is a nuclear-encoded mitochondrial matrix protein that stabilizes UQCRFS1 and chaperones it to the CIII complex. Defects in this gene are a cause of mitochondrial complex III deficiency, nuclear type 8. Three transcript variants encoding two different isoforms have been found for this gene.

Ubiquinol-cytochrome c reductase complex assembly factor 3 is a protein that in humans is encoded by the UQCC3 gene. Located in mitochondria, this protein is involved in the assembly of mitochondrial Complex III, stabilizing supercomplexes containing Complex III. Mutations in the UQCC3 gene cause Complex III deficiency with symptoms of hypoglycemia, hypotonia, lactic acidosis, and developmental delays. This protein plays an important role as an antiviral factor, bolstering the ability of cells to inhibit viral replication, independent of interferon production. The UQCC3 protein can be cleaved by OMA1 metalloprotease during mitochondrial depolarization, targeting the cell for apoptosis. Depletion of this protein alters cardiolipin composition, causing cellular and mitochondrial defects.
PET100 homolog is a protein that in humans is encoded by the PET100 gene. Mitochondrial complex IV, or cytochrome c oxidase, is a large transmembrane protein complex that is part of the respiratory electron transport chain of mitochondria. The small protein encoded by the PET100 gene plays a role in the biogenesis of mitochondrial complex IV. This protein localizes to the inner mitochondrial membrane and is exposed to the intermembrane space. Mutations in this gene are associated with mitochondrial complex IV deficiency. This gene has a pseudogene on chromosome 3. Alternative splicing results in multiple transcript variants.




