Viral evolution is a subfield of evolutionary biology and virology that is specifically concerned with the evolution of viruses. Viruses have short generation times, and many—in particular RNA viruses—have relatively high mutation rates. This elevated mutation rate, when combined with natural selection, allows viruses to quickly adapt to changes in their host environment. In addition, most viruses provide many offspring, so any mutated genes can be passed on to many offspring quickly. Although the chance of mutations and evolution can change depending on the type of virus, viruses overall have high chances for mutations.

Influenza A virus causes influenza in birds and some mammals, and is the only species of the genus Alphainfluenzavirus of the virus family Orthomyxoviridae. Strains of all subtypes of influenza A virus have been isolated from wild birds, although disease is uncommon. Some isolates of influenza A virus cause severe disease both in domestic poultry and, rarely, in humans. Occasionally, viruses are transmitted from wild aquatic birds to domestic poultry, and this may cause an outbreak or give rise to human influenza pandemics.
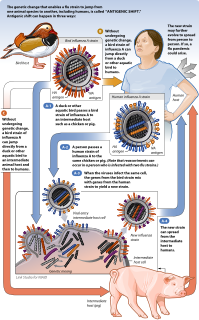
Antigenic shift is the process by which two or more different strains of a virus, or strains of two or more different viruses, combine to form a new subtype having a mixture of the surface antigens of the two or more original strains. The term is often applied specifically to influenza, as that is the best-known example, but the process is also known to occur with other viruses, such as visna virus in sheep. Antigenic shift is a specific case of reassortment or viral shift that confers a phenotypic change.

Orthomyxoviridae is a family of negative-sense RNA viruses. It includes seven genera: Alphainfluenzavirus, Betainfluenzavirus, Deltainfluenzavirus, Gammainfluenzavirus, Isavirus, Thogotovirus, and Quaranjavirus. The first four genera contain viruses that cause influenza in birds and mammals, including humans. Isaviruses infect salmon; the thogotoviruses are arboviruses, infecting vertebrates and invertebrates. The Quaranjaviruses are also arboviruses, infecting vertebrates (birds) and invertebrates (arthropods).
Antigenic drift is a kind of genetic variation in viruses, arising by the accumulation of mutations in the virus genes that code for virus-surface proteins that host antibodies recognize. This results in a new strain of virus particles that is not effectively inhibited by the antibodies that prevented infection by previous strains. This makes it easier for the changed virus to spread throughout a partially immune population. Antigenic drift occurs in both influenza A and influenza B viruses.

Influenza A virus subtype H5N1 (A/H5N1) is a subtype of the influenza A virus which can cause illness in humans and many other animal species. A bird-adapted strain of H5N1, called HPAI A(H5N1) for highly pathogenic avian influenza virus of type A of subtype H5N1, is the highly pathogenic causative agent of H5N1 flu, commonly known as avian influenza. It is enzootic in many bird populations, especially in Southeast Asia. One strain of HPAI A(H5N1) is spreading globally after first appearing in Asia. It is epizootic and panzootic, killing tens of millions of birds and spurring the culling of hundreds of millions of others to stem its spread. Many references to "bird flu" and H5N1 in the popular media refer to this strain.
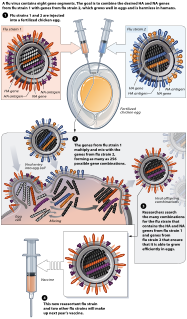
Reassortment is the mixing of the genetic material of a species into new combinations in different individuals. Several different processes contribute to reassortment, including assortment of chromosomes, and chromosomal crossover. It is particularly used when two similar viruses that are infecting the same cell exchange genetic material. In particular, reassortment occurs among influenza viruses, whose genomes consist of eight distinct segments of RNA. These segments act like mini-chromosomes, and each time a flu virus is assembled, it requires one copy of each segment.

Swine influenza is an infection caused by any one of several types of swine influenza viruses. Swine influenza virus (SIV) or swine-origin influenza virus (S-OIV) is any strain of the influenza family of viruses that is endemic in pigs. As of 2009, the known SIV strains include influenza C and the subtypes of influenza A known as H1N1, H1N2, H2N1, H3N1, H3N2, and H2N3.

Influenza A virus subtype H2N2 (A/H2N2) is a subtype of Influenza A virus. H2N2 has mutated into various strains including the "Asian flu" strain, H3N2, and various strains found in birds. It is also suspected of causing a human pandemic in 1889. The geographic spreading of the 1889 Russian flu has been studied and published.

Influenza A virus subtype H9N2 (A/H9N2) is a subtype of the species Influenza A virus.
An emergent virus is a virus that is either newly appeared, notably increasing in incidence/geographic range or has the potential to increase in the near future. Emergent viruses are a leading cause of emerging infectious diseases and raise public health challenges globally, given their potential to cause outbreaks of disease which can lead to epidemics and pandemics. As well as causing disease, emergent viruses can also have severe economic implications. Recent examples include the SARS-related coronaviruses, which have caused the 2002-2004 outbreak of SARS (SARS-CoV-1) and the 2019–20 pandemic of COVID-19 (SARS-CoV-2). Other examples include the human immunodeficiency virus which causes HIV/AIDS; the viruses responsible for Ebola; the H5N1 influenza virus responsible for avian flu; and H1N1/09, which caused the 2009 swine flu pandemic. Viral emergence in humans is often a consequence of zoonosis, which involves a cross-species jump of a viral disease into humans from other animals. As zoonotic viruses exist in animal reservoirs, they are much more difficult to eradicate and can therefore establish persistent infections in human populations.
H5N1 genetic structure is the molecular structure of the H5N1 virus's RNA.
Influenza research involves investigating molecular virology, pathogenesis, host immune responses, genomics, and epidemiology regarding influenza. The main goal of research is to develop influenza countermeasures such as vaccines, therapies and diagnostic tools.

Spanish flu research concerns studies regarding the causes and characteristics of the "Spanish flu," a variety of influenza that in 1918 was responsible for the worst influenza pandemic in modern history. Many theories about the origins and progress of the Spanish flu persisted in the literature, but it was not until 2005, when various samples of lung tissue were recovered from American World War I soldiers and from an Inupiat woman buried in permafrost in a mass grave in Brevig Mission, Alaska, that significant genetic research was made possible.
Antigenic variation or antigenic alteration refers to the mechanism by which an infectious agent such as a protozoan, bacterium or virus alters the proteins or carbohydrates on its surface and thus avoids a host immune response, making it one of the mechanisms of antigenic escape. It is related to phase variation. Antigenic variation not only enables the pathogen to avoid the immune response in its current host, but also allows re-infection of previously infected hosts. Immunity to re-infection is based on recognition of the antigens carried by the pathogen, which are "remembered" by the acquired immune response. If the pathogen's dominant antigen can be altered, the pathogen can then evade the host's acquired immune system. Antigenic variation can occur by altering a variety of surface molecules including proteins and carbohydrates. Antigenic variation can result from gene conversion, site-specific DNA inversions, hypermutation, or recombination of sequence cassettes. The result is that even a clonal population of pathogens expresses a heterogeneous phenotype. Many of the proteins known to show antigenic or phase variation are related to virulence.
Fujian flu refers to flu caused by either a Fujian human flu strain of the H3N2 subtype of the Influenza A virus or a Fujian bird flu strain of the H5N1 subtype of the Influenza A virus. These strains are named after Fujian, a coastal province in Southeast China.

Human mortality from H5N1 or the human fatality ratio from H5N1 or the case-fatality rate of H5N1 refer to the ratio of the number of confirmed human deaths resulting from confirmed cases of transmission and infection of H5N1 to the number of those confirmed cases. For example, if there are 100 confirmed cases of humans infected with H5N1 and 10 die, then there is a 10% human fatality ratio. H5N1 flu is a concern due to the global spread of H5N1 that constitutes a pandemic threat. The majority of H5N1 flu cases have been reported in southeast and east Asia. The case-fatality rate is central to pandemic planning. Estimates of case-fatality (CF) rates for past influenza pandemics have ranged from to 2-3% for the 1918 pandemic to about 0.6% for the 1957 pandemic to 0.2% for the 1968 pandemic. As of 2008, the official World Health Organization estimate for the case-fatality rate for the outbreak of H5N1 avian influenza was approximately 60%. Public health officials in Ontario, Canada argue that the true case-fatality rate could be lower, pointing to studies suggesting it could be 14–33%, but warned that it was unlikely to be as low as the 0.1–0.4% rate that was built into many pandemic plans.
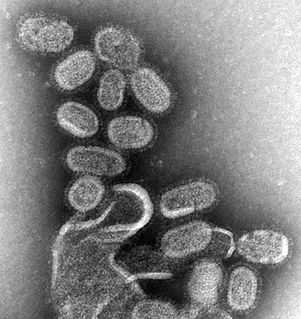
Influenza, commonly called "the flu", is an infectious disease caused by influenza viruses. Symptoms range from mild to severe and often include fever, runny nose, sore throat, muscle pain, headache, coughing, and fatigue. These symptoms typically begin 1–4 days after exposure to the virus and last for about 2–8 days. Diarrhea and vomiting can occur, particularly in children. Influenza may progress to pneumonia, which can be caused by the primary viral infection or by a secondary bacterial infection. Other complications of infection include acute respiratory distress syndrome, meningitis, encephalitis, and worsening of pre-existing health problems such as asthma and cardiovascular disease.
The pandemic H1N1/09 virus is a swine origin influenza A virus subtype H1N1 strain that was responsible for the 2009 swine flu pandemic. This strain is often called swine flu by the public media. For other names, see the Nomenclature section below.
Viral phylodynamics is defined as the study of how epidemiological, immunological, and evolutionary processes act and potentially interact to shape viral phylogenies. Since the coining of the term in 2004, research on viral phylodynamics has focused on transmission dynamics in an effort to shed light on how these dynamics impact viral genetic variation. Transmission dynamics can be considered at the level of cells within an infected host, individual hosts within a population, or entire populations of hosts.