 | |
| Clinical data | |
|---|---|
| Trade names | Buphenyl, Pheburane, Ammonaps, others |
| AHFS/Drugs.com | Micromedex Detailed Consumer Information |
| License data | |
| Pregnancy category |
|
| Routes of administration | By mouth |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Metabolism | Liver and kidney to phenylacetic acid |
| Elimination half-life | 0.8 hours (phenylbutyrate), 1.15-1.29 hours (phenylacetate) |
| Excretion | Urine (80-100% as phenylacetylglutamine) |
| Identifiers | |
| |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.130.318 |
| Chemical and physical data | |
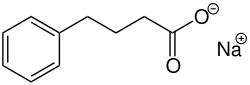
| Formula | C10H11NaO2 |
| Molar mass | 186.186 g·mol−1 |
| 3D model (JSmol) | |
| |
| |
| | |
Sodium phenylbutyrate, sold under the brand name Buphenyl among others, is a salt of an aromatic fatty acid, 4-phenylbutyrate (4-PBA) or 4-phenylbutyric acid. [7] The compound is used to treat urea cycle disorders, because its metabolites offer an alternative pathway to the urea cycle to allow excretion of excess nitrogen. [8] [9]
Contents
Sodium phenylbutyrate is also a histone deacetylase inhibitor and chemical chaperone, leading respectively to research into its use as an anti-cancer agent and in protein misfolding diseases such as cystic fibrosis. [7]
