G6PD is widely distributed in many species from bacteria to humans. Multiple sequence alignment of over 100 known G6PDs from different organisms reveal sequence identity ranging from 30% to 94%.[5] Human G6PD has over 30% identity in amino acid sequence to G6PD sequences from other species.[6] Humans also have two isoforms of a single gene coding for G6PD.[7] Moreover, at least 168 disease-causing mutations in this gene have been discovered.[8] These mutations are mainly missense mutations that result in amino acid substitutions,[9] and while some of them result in G6PD deficiency, others do not seem to result in any noticeable functional differences.[9] Some scientists have proposed that some of the genetic variation in human G6PD resulted from generations of adaptation to malarial infection.[10]
Substrate binding site of G6PD bound to G6P (shown in cream), from 2BHL. Phosphorus is shown in orange. Oxygen atoms of crystallographic waters are shown as red spheres. The conserved nine-peptide sequence of G6PD, and the partially conserved five-residue sequence of G6PD are shown in cyan and magenta respectively. All other amino acids from G6PD are shown in black. Hydrogen bonding and electrostatic interactions are shown by green dashed lines. All green dashes represent distances of less than 3.7 Å.
G6PD is generally found as a dimer of two identical monomers (see main thumbnail).[9] Depending on conditions, such as pH, these dimers can themselves dimerize to form tetramers.[6] Each monomer in the complex has a substrate binding site that binds to G6P, and a catalytic coenzyme binding site that binds to NADP+/NADPH using the Rossman fold.[5] For some higher organisms, such as humans, G6PD contains an additional NADP+ binding site, called the NADP+ structural site, that does not seem to participate directly in the reaction catalyzed by G6PD. The evolutionary purpose of the NADP+ structural site is unknown.[5] As for size, each monomer is approximately 500 amino acids long (514 amino acids for humans[6]).
Functional and structural conservation between human G6PD and Leuconostoc mesenteroides G6PD points to three widely conserved regions on the enzyme: a nine-residue peptide in the substrate binding site, RIDHYLGKE (residues 198–206 on human G6PD), a nucleotide-binding fingerprint, GxxGDLA (residues 38–44 on human G6PD), and a partially conserved sequence EKPxG near the substrate binding site (residues 170–174 on human G6PD), where we have use "x" to denote a variable amino acid.[5] The crystal structure of G6PD reveals an extensive network of electrostatic interactions and hydrogen bonding involving G6P, three water molecules, three lysine residues, one arginine, two histidines, two glutamic acids, and other polar amino acids.
The proline at position 172 is thought to play a crucial role in positioning Lys171 correctly with respect to the substrate, G6P. In the two crystal structures of normal human G6P, Pro172 is seen exclusively in the cis conformation, while in the crystal structure of one disease causing mutant (variant Canton R459L), Pro172 is seen almost exclusively in the trans conformation.[5]
With access to crystal structures, some scientists have tried to model the structures of other mutants. For example, in German ancestry, where enzymopathy due to G6PD deficiency is rare, mutation sites on G6PD have been shown to lie near the NADP+ binding site, the G6P binding site, and near the interface between the two monomers. Thus, mutations in these critical areas are possible without completely disrupting the function of G6PD.[9] In fact, it has been shown that most disease causing mutations of G6PD occur near the NADP+ structural site.[14]
The NADP+ structural site is located greater than 20Å away from the substrate binding site and the catalytic coenzyme NADP+ binding site. Its purpose in the enzyme catalyzed reaction has been unclear for many years. For some time, it was thought that NADP+ binding to the structural site was necessary for dimerization of the enzyme monomers. However, this was shown to be incorrect.[14] On the other hand, it was shown that the presence of NADP+ at the structural site promotes the dimerization of dimers to form enzyme tetramers.[14] It was also thought that the tetramer state was necessary for catalytic activity; however, this too was shown to be false.[14] The NADP+ structural site is quite different from the NADP+ catalytic coenzyme binding site, and contains the nucleotide-binding fingerprint.
The structural site bound to NADP+ possesses favorable interactions that keep it tightly bound. In particular, there is a strong network of hydrogen bonding with electrostatic charges being diffused across multiple atoms through hydrogen bonding with four water molecules (see figure). Moreover, there is an extremely strong set of hydrophobic stacking interactions that result in overlapping π systems.
Hydrogen bonding and electrostatic interaction network (green). All green dashes represent distances less than 3.8 Å
Hydrophobic stacking interactions (green). All green dashes represent distances less than 4.4 Å. Slightly different view than the first panel.
NADP+ structural site of G6PD. NADP+ is shown in cream. Phosphorus is shown in orange. The oxygen atoms of crystallographic water molecules are shown as red spheres. The conserved 9-peptide sequence of G6PD is show in cyan.
The structural site has been shown to be important for maintaining the long term stability of the enzyme.[14] More than 40 severe class I mutations involve mutations near the structural site, thus affecting the long term stability of these enzymes in the body, ultimately resulting in G6PD deficiency.[14] For example, two severe class I mutations, G488S and G488V, drastically increase the dissociation constant between NADP+ and the structural site by a factor of 7 to 13. With the proximity of residue 488 to Arg487, it is thought that a mutation at position 488 could affect the positioning of Arg487 relative to NADP+,[14] and thus disrupt binding.
Regulation
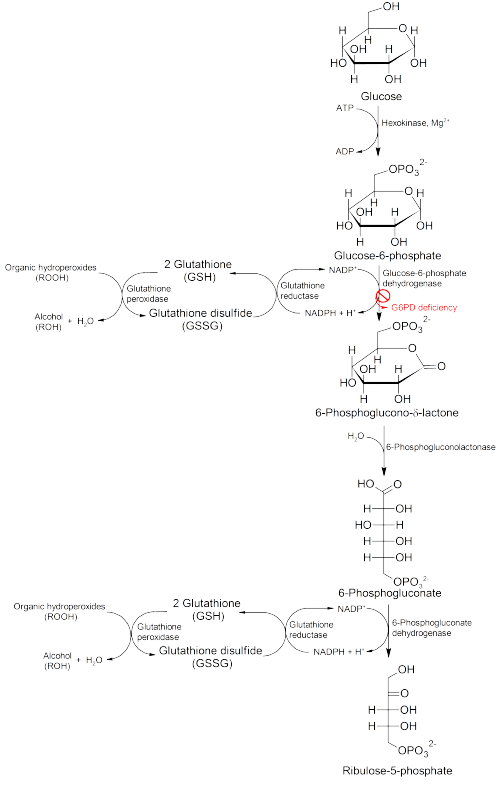
G6PD converts G6P into 6-phosphoglucono-δ-lactone and is the rate-limiting enzyme of the pentose phosphate pathway. Thus, regulation of G6PD has downstream consequences for the activity of the rest of the pentose phosphate pathway.
Glucose-6-phosphate dehydrogenase is stimulated by its substrate G6P. The usual ratio of NADPH/NADP+ in the cytosol of tissues engaged in biosyntheses is about 100/1. Increased utilization of NADPH for fatty acid biosynthesis will dramatically increase the level of NADP+, thus stimulating G6PD to produce more NADPH. Yeast G6PD is inhibited by long chain fatty acids according to two older publications[15][16] and might be product inhibition in fatty acid synthesis which requires NADPH.
G6PD is negatively regulated by acetylation on lysine 403 (Lys403), an evolutionarily conserved residue. The K403 acetylated G6PD is incapable of forming active dimers and displays a complete loss of activity. Mechanistically, acetylating Lys403 sterically hinders the NADP+ from entering the NADP+ structural site, which reduces the stability of the enzyme. Cells sense extracellular oxidative stimuli to decrease G6PD acetylation in a SIRT2-dependent manner. The SIRT2-mediated deacetylation and activation of G6PD stimulates pentose phosphate pathway to supply cytosolic NADPH to counteract oxidative damage and protect mouse erythrocytes.[17]
G6PD is remarkable for its genetic diversity. Many variants of G6PD, mostly produced from missense mutations, have been described with wide-ranging levels of enzyme activity and associated clinical symptoms. Two transcript variants encoding different isoforms have been found for this gene.[20]
Cell growth and proliferation are affected by G6PD.[21] Pharmacologically ablating G6PD has been shown to overcome cross-tolerance of breast cancer cells to anthracyclines.[22] G6PD inhibitors are under investigation to treat cancers and other conditions.[19]In vitro cell proliferation assay indicates that G6PD inhibitors, DHEA (dehydroepiandrosterone) and ANAD (6-aminonicotinamide), effectively decrease the growth of AML cell lines.[21][23] G6PD is hypomethylated at K403 in acute myeloid leukemia, SIRT2 activates G6PD to enhance NADPH production and promote leukemia cell proliferation.[23]
↑Eger-Neufeldt I, Teinzer A, Weiss L, Wieland O (March 1965). "Inhibition of glucose-6-phosphate dehydrogenase by long chain acyl-coenzyme A". Biochemical and Biophysical Research Communications. 19 (1): 43–48. Bibcode:1965BBRC...19...43E. doi:10.1016/0006-291X(65)90116-6.
Wajcman H, Galactéros F (August 2004). "[Glucose 6-phosphate dehydrogenase deficiency: a protection against malaria and a risk for hemolytic accidents]". Comptes Rendus Biologies (in French). 327 (8): 711–20. doi:10.1016/j.crvi.2004.07.010. PMID15506519.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.