Sigma receptors (σ-receptors) are protein receptors that bind ligands such as 4-PPBP, SA 4503 (cutamesine), ditolylguanidine, dimethyltryptamine, and siramesine. There are two subtypes, sigma-1 receptors (σ1) and sigma-2 receptors (σ2), which are classified as sigma receptors for their pharmacological similarities, even though they are evolutionarily unrelated.
Melanocortin receptors are members of the rhodopsin family of 7-transmembrane G protein-coupled receptors.

The sigma-1 receptor (σ1R), one of two sigma receptor subtypes, is a chaperone protein at the endoplasmic reticulum (ER) that modulates calcium signaling through the IP3 receptor. In humans, the σ1 receptor is encoded by the SIGMAR1 gene.

The δ-opioid receptor, also known as delta opioid receptor or simply delta receptor, abbreviated DOR or DOP, is an inhibitory 7-transmembrane G-protein coupled receptor coupled to the G protein Gi/G0 and has enkephalins as its endogenous ligands. The regions of the brain where the δ-opioid receptor is largely expressed vary from species model to species model. In humans, the δ-opioid receptor is most heavily expressed in the basal ganglia and neocortical regions of the brain.
Cutamesine (SA 4503) is a synthetic sigma receptor agonist which is selective for the σ1 receptor, a chaperone protein mainly found in the endoplasmic reticulum of cells in the central nervous system. These σ1 receptors play a key role in the modulation of Ca2+ release and apoptosis. Cutamesine's activation of the σ1 receptor is tied to a variety of physiological phenomena in the CNS, including activation of dopamine-releasing neurons and repression of the MAPK/ERK pathway.

Siramesine is a sigma receptor agonist, selective for the σ2 subtype. In animal studies, siramesine has been shown to produce anxiolytic and antidepressant effects. It was developed by the pharmaceutical company H Lundbeck for the treatment of anxiety, although development was discontinued after clinical trials showed a lack of efficacy in humans. Siramesine has been shown to produce an enhanced antidepressant effect when co-administered with NMDA antagonists. It has also been used to study the σ2 activity of cocaine, and has been shown to produce anticancer properties both in vitro and in vivo.

The sigma-2 receptor (σ2R) is a sigma receptor subtype that has attracted attention due to its involvement in diseases such as neurological diseases, neurodegenerative, neuro-ophthalmic and cancer. It is currently under investigation for its potential diagnostic and therapeutic uses.

Magnolol is an organic compound that is classified as lignan. It is a bioactive compound found in the bark of the Houpu magnolia and in M. grandiflora. The compound exists at the level of a few percent in the bark of species of magnolia, the extracts of which have been used in traditional Chinese and Japanese medicine. In addition to magnolol, related lignans occur in the extracts including honokiol, which is an isomer of magnolol.

The human muscarinic acetylcholine receptor M5, encoded by the CHRM5 gene, is a member of the G protein-coupled receptor superfamily of integral membrane proteins. It is coupled to Gq protein. Binding of the endogenous ligand acetylcholine to the M5 receptor triggers a number of cellular responses such as adenylate cyclase inhibition, phosphoinositide degradation, and potassium channel modulation. Muscarinic receptors mediate many of the effects of acetylcholine in the central and peripheral nervous system. The clinical implications of this receptor have not been fully explored; however, stimulation of this receptor is known to effectively decrease cyclic AMP levels and downregulate the activity of protein kinase A (PKA).

The adenosine A2B receptor, also known as ADORA2B, is a G-protein coupled adenosine receptor, and also denotes the human adenosine A2b receptor gene which encodes it.

Dopamine receptor D3 is a protein that in humans is encoded by the DRD3 gene.

The 5-HT7 receptor is a member of the GPCR superfamily of cell surface receptors and is activated by the neurotransmitter serotonin (5-hydroxytryptamine, 5-HT). The 5-HT7 receptor is coupled to Gs (stimulates the production of the intracellular signaling molecule cAMP) and is expressed in a variety of human tissues, particularly in the brain, the gastrointestinal tract, and in various blood vessels. This receptor has been a drug development target for the treatment of several clinical disorders. The 5-HT7 receptor is encoded by the HTR7 gene, which in humans is transcribed into 3 different splice variants.
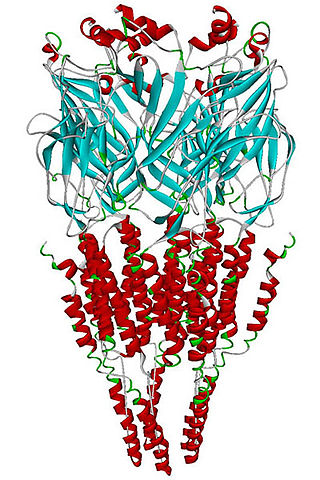
The alpha-7 nicotinic receptor, also known as the α7 receptor, is a type of nicotinic acetylcholine receptor implicated in long-term memory, consisting entirely of α7 subunits. As with other nicotinic acetylcholine receptors, functional α7 receptors are pentameric [i.e., (α7)5 stoichiometry].
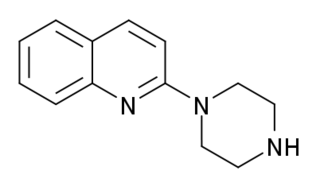
Quipazine, also known as 1-(2-quinolinyl)piperazine, is a serotonergic drug of the arylpiperazine family and an analogue of 1-(2-pyridinyl)piperazine which is used in scientific research. It was first described in the 1960s and was originally intended as an antidepressant but was never developed or marketed for medical use.
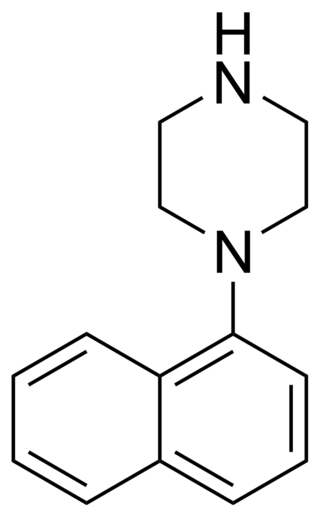
1-(1-Naphthyl)piperazine (1-NP) is a drug which is a phenylpiperazine derivative.
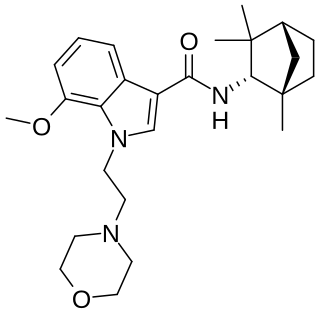
MN-25 (UR-12) is a drug invented by Bristol-Myers Squibb, that acts as a reasonably selective agonist of peripheral cannabinoid receptors. It has moderate affinity for CB2 receptors with a Ki of 11 nM, but 22x lower affinity for the psychoactive CB1 receptors with a Ki of 245 nM. The indole 2-methyl derivative has the ratio of affinities reversed however, with a Ki of 8 nM at CB1 and 29 nM at CB2, which contrasts with the usual trend of 2-methyl derivatives having increased selectivity for CB2 (cf. JWH-018 vs JWH-007, JWH-081 vs JWH-098).
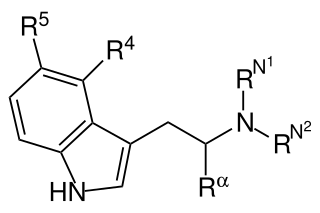
Substituted tryptamines, or simply tryptamines, also known as serotonin analogues (i.e., 5-hydroxytryptamine analogues), are organic compounds which may be thought of as being derived from tryptamine itself. The molecular structures of all tryptamines contain an indole ring, joined to an amino (NH2) group via an ethyl (−CH2–CH2−) sidechain. In substituted tryptamines, the indole ring, sidechain, and/or amino group are modified by substituting another group for one of the hydrogen (H) atoms.

N-Arachidonylglycine (NAGly) is a carboxylic metabolite of the endocannabinoid anandamide (AEA). Since it was first synthesized in 1996, NAGly has been a primary focus of the relatively contemporary field of lipidomics due to its wide range of signaling targets in the brain, the immune system and throughout various other bodily systems. In combination with 2‐arachidonoyl glycerol (2‐AG), NAGly has enabled the identification of a family of lipids often referred to as endocannabinoids. Recently, NAGly has been found to bind to G-protein coupled receptor 18 (GPR18), the putative abnormal cannabidiol receptor. NaGly is an endogenous inhibitor of fatty acid amide hydrolase (FAAH) and thereby increases the ethanolamide endocannabinoids AEA, oleoylethanolamide (OEA) and palmitoylethanolamide (PEA) levels. NaGly is found throughout the body and research on its explicit functions is ongoing.
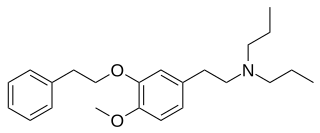
NE-100 or 4-methoxy-3-(2-phenylethoxy)-N,N-dipropylbenzeneethanamine is a selective sigma-1 receptor antagonist, with a reported binding affinity of Ki = 1.03 ± 0.01 nM, and more than 205 times selectivity over the sigma-2 receptor.
