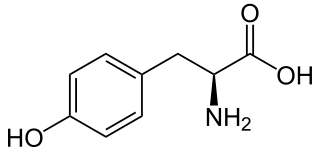
Tyrosine or 4-hydroxyphenylalanine is one of the 20 standard amino acids that are used by cells to synthesize proteins. It is a non-essential amino acid with a polar side group. The word "tyrosine" is from the Greek tyrós, meaning cheese, as it was first discovered in 1846 by German chemist Justus von Liebig in the protein casein from cheese. It is called tyrosyl when referred to as a functional group or side chain. While tyrosine is generally classified as a hydrophobic amino acid, it is more hydrophilic than phenylalanine. It is encoded by the codons UAC and UAU in messenger RNA.

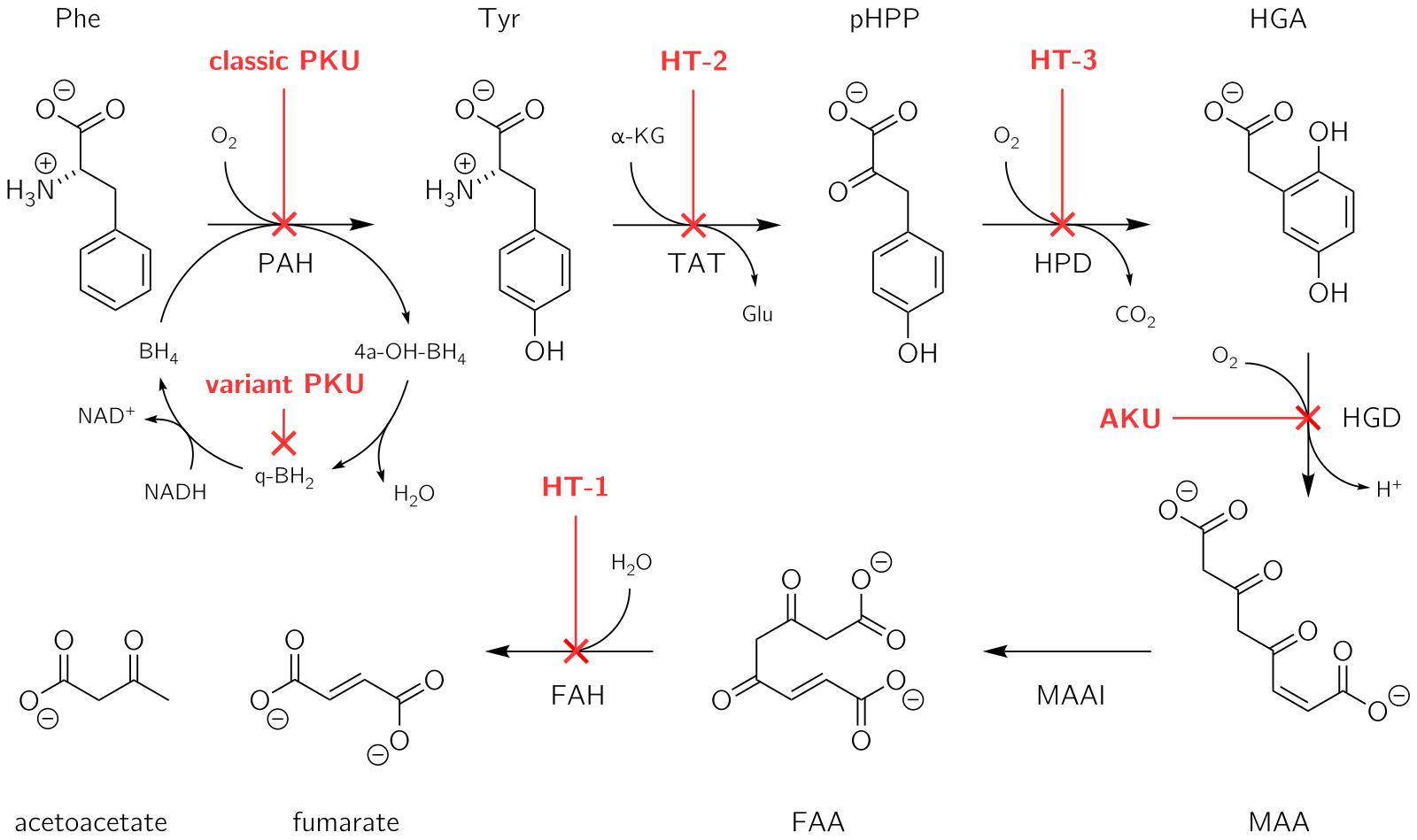
Alkaptonuria is a rare inherited genetic disorder in which the body cannot process the amino acids phenylalanine and tyrosine, which occur in protein. It is caused by a mutation in the HGD gene for the enzyme homogentisate 1,2-dioxygenase ; if a person inherits an abnormal copy from both parents, the body accumulates an intermediate substance called homogentisic acid in the blood and tissues. Homogentisic acid and its oxidized form alkapton are excreted in the urine, giving it an unusually dark color. The accumulating homogentisic acid causes damage to cartilage and heart valves, as well as precipitating as kidney stones and stones in other organs. Symptoms usually develop in people over 30 years old, although the dark discoloration of the urine is present from birth.

A glycogen storage disease is a metabolic disorder caused by enzyme deficiencies affecting either glycogen synthesis, glycogen breakdown or glycolysis, typically in muscles and/or liver cells.

Hyperammonemia is a metabolic disturbance characterised by an excess of ammonia in the blood. It is a dangerous condition that may lead to brain injury and death. It may be primary or secondary.

Angiotensin-converting enzyme, or ACE, is a central component of the renin–angiotensin system (RAS), which controls blood pressure by regulating the volume of fluids in the body. It converts the hormone angiotensin I to the active vasoconstrictor angiotensin II. Therefore, ACE indirectly increases blood pressure by causing blood vessels to constrict. ACE inhibitors are widely used as pharmaceutical drugs for treatment of cardiovascular diseases.
Inborn errors of metabolism form a large class of genetic diseases involving congenital disorders of metabolism. The majority are due to defects of single genes that code for enzymes that facilitate conversion of various substances (substrates) into others (products). In most of the disorders, problems arise due to accumulation of substances which are toxic or interfere with normal function, or to the effects of reduced ability to synthesize essential compounds. Inborn errors of metabolism are now often referred to as congenital metabolic diseases or inherited metabolic disorders. The term inborn errors of metabolism was coined by a British physician, Archibald Garrod (1857–1936), in 1908. He is known for work that prefigured the "one gene-one enzyme" hypothesis, based on his studies on the nature and inheritance of alkaptonuria. His seminal text, Inborn Errors of Metabolism was published in 1923.

Fumonisin B1 is the most prevalent member of a family of toxins, known as fumonisins, produced by several species of Fusarium molds, such as Fusarium verticillioides, which occur mainly in maize (corn), wheat and other cereals. Fumonisin B1 contamination of maize has been reported worldwide at mg/kg levels. Human exposure occurs at levels of micrograms to milligrams per day and is greatest in regions where maize products are the dietary staple.

Tyrosinemia or tyrosinaemia is an error of metabolism, usually inborn, in which the body cannot effectively break down the amino acid tyrosine. Symptoms of untreated tyrosinemia include liver and kidney disturbances. Without treatment, tyrosinemia leads to liver failure. Today, tyrosinemia is increasingly detected on newborn screening tests before any symptoms appear. With early and lifelong management involving a low-protein diet, special protein formula, and sometimes medication, people with tyrosinemia develop normally, are healthy, and live normal lives.

4-Hydroxyphenylpyruvate dioxygenase (HPPD), also known as α-ketoisocaproate dioxygenase, is an Fe(II)-containing non-heme oxygenase that catalyzes the second reaction in the catabolism of tyrosine - the conversion of 4-hydroxyphenylpyruvate into homogentisate. HPPD also catalyzes the conversion of phenylpyruvate to 2-hydroxyphenylacetate and the conversion of α-ketoisocaproate to β-hydroxy β-methylbutyrate. HPPD is an enzyme that is found in nearly all aerobic forms of life.
The epoxyeicosatrienoic acids or EETs are signaling molecules formed within various types of cells by the metabolism of arachidonic acid by a specific subset of Cytochrome P450 enzymes termed cytochrome P450 epoxygenases. These nonclassic eicosanoids are generally short-lived, being rapidly converted from epoxides to less active or inactive dihydroxy-eicosatrienoic acids (diHETrEs) by a widely distributed cellular enzyme, Soluble epoxide hydrolase (sEH), also termed Epoxide hydrolase 2. The EETs consequently function as transiently acting, short-range hormones; that is, they work locally to regulate the function of the cells that produce them or of nearby cells. The EETs have been most studied in animal models where they show the ability to lower blood pressure possibly by a) stimulating arterial vasorelaxation and b) inhibiting the kidney's retention of salts and water to decrease intravascular blood volume. In these models, EETs prevent arterial occlusive diseases such as heart attacks and brain strokes not only by their anti-hypertension action but possibly also by their anti-inflammatory effects on blood vessels, their inhibition of platelet activation and thereby blood clotting, and/or their promotion of pro-fibrinolytic removal of blood clots. With respect to their effects on the heart, the EETs are often termed cardio-protective. Beyond these cardiovascular actions that may prevent various cardiovascular diseases, studies have implicated the EETs in the pathological growth of certain types of cancer and in the physiological and possibly pathological perception of neuropathic pain. While studies to date imply that the EETs, EET-forming epoxygenases, and EET-inactivating sEH can be manipulated to control a wide range of human diseases, clinical studies have yet to prove this. Determination of the role of the EETS in human diseases is made particularly difficult because of the large number of EET-forming epoxygenases, large number of epoxygenase substrates other than arachidonic acid, and the large number of activities, some of which may be pathological or injurious, that the EETs possess.

Hawkinsinuria, is an autosomal dominant metabolic disorder affecting the metabolism of tyrosine.

Nitisinone (INN), also known as NTBC is a medication used to slow the effects of hereditary tyrosinemia type 1 (HT-1).

Essential fructosuria, caused by a deficiency of the enzyme hepatic fructokinase, is a clinically benign condition characterized by the incomplete metabolism of fructose in the liver, leading to its excretion in urine. Fructokinase is the first enzyme involved in the degradation of fructose to fructose-1-phosphate in the liver. This defective degradation does not cause any clinical symptoms, fructose is either excreted unchanged in the urine or metabolized to fructose-6-phosphate by alternate pathways in the body, most commonly by hexokinase in adipose tissue and muscle.

Tyrosine aminotransferase is an enzyme present in the liver and catalyzes the conversion of tyrosine to 4-hydroxyphenylpyruvate.

Fumarylacetoacetase is an enzyme that in humans is encoded by the FAH gene located on chromosome 15. The FAH gene is thought to be involved in the catabolism of the amino acid phenylalanine in humans.

Phenylacetylglutamine is a product formed by the conjugation of phenylacetate and glutamine. It is a common metabolite that is naturally occurring in human urine.

In enzymology, maleylacetoacetate isomerase is an enzyme that catalyzes the chemical reaction
Tyrosinemia type III is a rare disorder caused by a deficiency of the enzyme 4-hydroxyphenylpyruvate dioxygenase, encoded by the gene HPD. This enzyme is abundant in the liver, and smaller amounts are found in the kidneys. It is one of a series of enzymes needed to break down tyrosine. Specifically, 4-hydroxyphenylpyruvate dioxygenase converts a tyrosine byproduct called 4-hydroxyphenylpyruvate to homogentisic acid. Characteristic features of type III tyrosinemia include mild mental retardation, seizures, and periodic loss of balance and coordination. Type III tyrosinemia is very rare; only a few cases have been reported.
Tyrosinemia type II is an autosomal recessive condition with onset between ages 2 and 4 years, when painful circumscribed calluses develop on the pressure points of the palm of the hand and sole of the foot.
4-Hydroxyphenylpyruvate dioxygenase (HPPD) inhibitors are a class of herbicides that prevent plants by blocking 4-Hydroxyphenylpyruvate dioxygenase, an enzyme in plants that breaks down the amino acid tyrosine into molecules that are then used by plants to create other molecules that plants need. This process of breakdown, or catabolism, and making new molecules from the results, or biosynthesis, is something all living things do. HPPD inhibitors were first brought to market in 1980, although their mechanism of action was not understood until the late 1990s. They were originally used primarily in Japan in rice production, but since the late 1990s have been used in Europe and North America for corn, soybeans, and cereals, and since the 2000s have become more important as weeds have become resistant to glyphosate and other herbicides. Genetically modified crops are under development that include resistance to HPPD inhibitors. There is a pharmaceutical drug on the market, nitisinone, that was originally under development as an herbicide as a member of this class, and is used to treat an orphan disease, type I tyrosinemia.