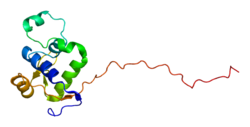
Structure
WRN is a member of the RecQ Helicase family. It is the only RecQ Helicase that contains 3' to 5' exonuclease activity. WRN is an oligomer that can act as a monomer when unwinding DNA, but as a dimer in solution or a tetramer when complexed with DNA, and has also been observed in hexameric forms. The diffusion of WRN has been measured to 1.62  in nucleoplasm and 0.12
in nucleoplasm and 0.12  at nucleoli. [6]
at nucleoli. [6]
Orthologs of WRN have been found in a number of other organisms, including Drosophila, Xenopus , and C. elegans. The amino terminus of WRN is involved in both helicase and nuclease activities, while the carboxyl-terminus interacts with p53, an important tumor suppressor. [7] mRNA that codes for WRN has been identified in most human tissues. [7]
Post-translational modification
Phosphorylation of WRN at serine/threonine inhibits helicase and exonuclease activities which are important to post-replication DNA repair. De-phosphorylation at these sites enhances the catalytic activities of WRN. Phosphorylation may affect other post-translational modifications, including SUMOylation and acetylation. [8] Upon its inhibition by a small molecule in cancer cells harboring a high number of microsatellites (MSI-H), WRN becomes SUMOylated, which leads to is ubiquitylation and subsequent degradation. [9]
Methylation of WRN causes the gene to turn off. This suppresses the production of the WRN protein and its functions in DNA repair. [10]
Function
The exonuclease activities of WRN include degradation of recessed 3' ends and initiation of DNA degradation from a gap in dsDNA. WRN is important in repair of double strand breaks by homologous recombination [11] [12] or non-homologous end joining, [13] repair of single nucleotide damages by base excision repair, [14] [15] [5] and is effective in replication arrest recovery. [16]
WRN may also be important in telomere maintenance and replication, especially the replication of the G-rich sequences. [8]
WRN may function as an exonuclease in DNA repair, recombination, or replication, as well as resolution of DNA secondary structures. It is involved in branch migration at Holliday junctions, and it interacts with other DNA replication intermediates. [16] WRN is important to genome stability, and cells with mutations to WRN are more susceptible to DNA damage and DNA breaks. [17]
Homologous recombinational repair
WRN is active in homologous recombination. Cells defective in the WRN gene have a 23-fold reduction in spontaneous mitotic recombination, with especial deficiency in conversion-type events. [18] WRN defective cells, when exposed to x-rays, have more chromosome breaks and micronuclei than cells with wild-type WRN. [19] Cells defective in the WRN gene are not more sensitive than wild-type cells to gamma-irradiation, UV light, 4 – 6 cyclobutane pyrimidines, or mitomycin C, but are sensitive to type I and type II topoisomerase inhibitors. [20] These findings suggested that the WRN protein takes part in homologous recombinational repair and in the processing of stalled replication forks. [21]
Non-homologous end joining
WRN has an important role in non-homologous end joining (NHEJ) DNA repair. As shown by Shamanna et al., [13] WRN is recruited to double-strand breaks (DSBs) and participates in NHEJ with its enzymatic and non-enzymatic functions. At DSBs, in association with Ku (protein), it promotes standard or canonical NHEJ (c-NHEJ), repairing double-strand breaks in DNA with its enzymatic functions and with a fair degree of accuracy. WRN inhibits an alternative form of NHEJ, called alt-NHEJ or microhomology-mediated end joining (MMEJ). MMEJ is an inaccurate mode of repair for double-strand breaks.
Base excision repair
WRN has a role in base excision repair (BER) of DNA. As shown by Das et al., [14] WRN associates with NEIL1 in the early damage-sensing step of BER. WRN stimulates NEIL1 in excision of oxidative lesions. NEIL1 is a DNA glycosylase that initiates the first step in BER by cleaving bases damaged by reactive oxygen species (ROS) and introducing a DNA strand break via NEIL1's associated lyase activity. [22] NEIL1 recognizes (targets) and removes certain ROS-damaged bases and then incises the abasic site via β,δ elimination, leaving 3′ and 5′ phosphate ends. NEIL1 recognizes oxidized pyrimidines, formamidopyrimidines, thymine residues oxidized at the methyl group, and both stereoisomers of thymine glycol. [23]
WRN also participates in BER through its interaction with Polλ. [15] WRN binds to the catalytic domain of Polλ and specifically stimulates DNA gap filling by Polλ over 8-oxo-G followed by strand displacement synthesis. This allows WRN to promote long-patch DNA repair synthesis by Polλ during MUTYH-initiated repair of 8-oxo-G:A mispairs.
Replication arrest recovery
WRN is also involved in replication arrest recovery. If WRN is defective, replication arrest results in accumulation of DSBs and enhanced chromosome fragmentation. [24] As shown by Pichierri et al., [24] WRN interacts with the RAD9-RAD1-HUS1 (9.1.1) complex, one of the central factors of the replication checkpoint. This interaction is mediated by the binding of the RAD1 subunit to the N-terminal region of WRN and is instrumental for WRN relocalization to nuclear foci and its phosphorylation in response to replication arrest. (In the absence of DNA damage or replication fork stalling, WRN protein remains localized to the nucleoli. [25] ) The interaction of WRN with the 9.1.1 complex results in prevention of DSB formation at stalled replication forks. [24]
Apoptosis
The p53 protein and WRN helicase engage in direct protein-protein interaction. [26] Increased cellular WRN levels elicit increased cellular p53 levels and also potentiate p53-mediated apoptosis. [26] This finding suggests that WRN helicase participates in the activation of p53 in response to certain types of DNA damage. [26] p53-mediated apoptosis is attenuated in cells from patients with Werner syndrome. [27]
Both repair of DNA damage and apoptosis are enzymatic processes necessary for maintaining integrity of the genome in humans. Cells with insufficient DNA repair tend to accumulate DNA damages, and when such cells are also defective in apoptosis they tend to survive even though excessive DNA damages are present. [28] Replication of DNA in such deficient cells tends to lead to mutations and such mutations may cause cancer. Thus Werner syndrome helicase appears to have two roles related to the prevention of cancer, where the first role is to promote repair of specific types of damage and the second role is to induce apoptosis if the level of such DNA damage is beyond the cell's repair capability [28]
This page is based on this
Wikipedia article Text is available under the
CC BY-SA 4.0 license; additional terms may apply.
Images, videos and audio are available under their respective licenses.