
Fluvoxamine, commonly sold under the brand names Luvox and Faverin, is an antidepressant of the selective serotonin reuptake inhibitor (SSRI) class. It is primarily used to treat major depressive disorder and obsessive–compulsive disorder (OCD), but is also used to treat anxiety disorders such as panic disorder, social anxiety disorder, and post-traumatic stress disorder.

Hydroxyzine, sold under the brand names Atarax and Vistaril among others, is an antihistamine medication. It is used in the treatment of itchiness, insomnia, anxiety, and nausea, including that due to motion sickness. It is used either by mouth or injection into a muscle.

Buspirone, sold under the brand name Buspar, among others, is an anxiolytic, a medication primarily used to treat anxiety disorders, particularly generalized anxiety disorder. It is a serotonin 5-HT1A receptor agonist, increasing action at serotonin receptors in the brain. It is taken orally, and takes two to six weeks to be fully effective.

Cetirizine is a second-generation antihistamine used to treat allergic rhinitis, dermatitis, and urticaria (hives). It is taken by mouth. Effects generally begin within thirty minutes and last for about a day. The degree of benefit is similar to other antihistamines such as diphenhydramine, which is a first-generation antihistamine.

Zopiclone, sold under the brand name Imovane among others, is a nonbenzodiazepine used to treat difficulty sleeping. Zopiclone is molecularly distinct from benzodiazepine drugs and is classed as a cyclopyrrolone. However, zopiclone increases the normal transmission of the neurotransmitter gamma-aminobutyric acid (GABA) in the central nervous system, via modulating GABAA receptors similarly to the way benzodiazepine drugs do.

Doxepin is a medication belonging to the tricyclic antidepressant (TCA) class of drugs used to treat major depressive disorder, anxiety disorders, chronic hives, and insomnia. For hives it is a less preferred alternative to antihistamines. It has a mild to moderate benefit for sleeping problems. It is used as a cream for itchiness due to atopic dermatitis or lichen simplex chronicus.

Alpidem, sold under the brand name Ananxyl, is a nonbenzodiazepine anxiolytic medication which was briefly used to treat anxiety disorders but is no longer marketed. It was previously marketed in France, but was discontinued due to liver toxicity. Alpidem is taken by mouth.

Prazepam is a benzodiazepine derivative drug developed by Warner-Lambert in the 1960s. It possesses anxiolytic, anticonvulsant, sedative and skeletal muscle relaxant properties. Prazepam is a prodrug for desmethyldiazepam which is responsible for the therapeutic effects of prazepam.

Camazepam is a benzodiazepine psychoactive drug, marketed under the brand names Albego, Limpidon and Paxor. It is the dimethyl carbamate ester of temazepam, a metabolite of diazepam. While it possesses anxiolytic, anticonvulsant, skeletal muscle relaxant and hypnotic properties it differs from other benzodiazepines in that its anxiolytic properties are particularly prominent but has comparatively limited anticonvulsant, hypnotic and skeletal muscle relaxant properties.
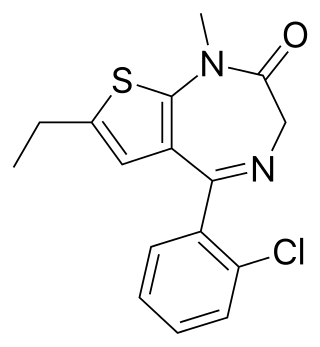
Clotiazepam is a thienodiazepine drug which is a benzodiazepine analog. The clotiazepam molecule differs from benzodiazepines in that the benzene ring has been replaced by a thiophene ring. It possesses anxiolytic, skeletal muscle relaxant, anticonvulsant, sedative properties. Stage 2 NREM sleep is significantly increased by clotiazepam.
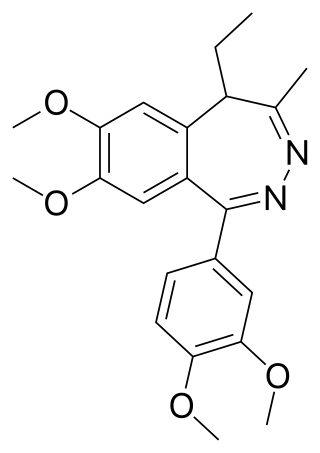
Tofisopam is an anxiolytic that is marketed in several European countries. Chemically, it is a 2,3-benzodiazepine. Unlike other anxiolytic benzodiazepines however, tofisopam does not have anticonvulsant, sedative, skeletal muscle relaxant, motor skill-impairing or amnestic properties. While it may not be an anticonvulsant in and of itself, it has been shown to enhance the anticonvulsant action of classical 1,4-benzodiazepines and muscimol, but not sodium valproate, carbamazepine, phenobarbital, or phenytoin. Tofisopam is indicated for the treatment of anxiety and alcohol withdrawal, and is prescribed in a dosage of 50–300 mg per day divided into three doses. Peak plasma levels are attained two hours after an oral dose. Tofisopam is not reported as causing dependence to the same extent as other benzodiazepines, but is still recommended to be prescribed for a maximum of 12 weeks.

Delorazepam, also known as chlordesmethyldiazepam and nordiclazepam, is a drug which is a benzodiazepine and a derivative of desmethyldiazepam. It is marketed in Italy, where it is available under the trade name EN and Dadumir. Delorazepam (chlordesmethyldiazepam) is also an active metabolite of the benzodiazepine drugs diclazepam and cloxazolam. Adverse effects may include hangover type effects, drowsiness, behavioural impairments and short-term memory impairments. Similar to other benzodiazepines delorazepam has anxiolytic, skeletal muscle relaxant, hypnotic and anticonvulsant properties.

Benzoctamine is a drug that possesses sedative and anxiolytic properties. Marketed as Tacitin by Ciba-Geigy, it is different from most sedative drugs because in most clinical trials it does not produce respiratory depression, but actually stimulates the respiratory system. As a result, when compared to other sedative and anxiolytic drugs such as benzodiazepines like diazepam, it is a safer form of tranquilizing. However, when co-administered with other drugs that cause respiratory depression, like morphine, it can cause increased respiratory depression.

Metaclazepam is a drug which is a benzodiazepine derivative. It is a relatively selective anxiolytic with less sedative or muscle relaxant properties than other benzodiazepines such as diazepam or bromazepam. It has an active metabolite N-desmethylmetaclazepam, which is the main metabolite of metaclazepam. There is no significant difference in metabolism between younger and older individuals.

Antalarmin (CP-156,181) is a drug that acts as a CRH1 antagonist.

Premazepam is a Pyrrolodiazepine class of drug. It is a partial agonist of benzodiazepine receptors and was shown in 1984 to possess both anxiolytic and sedative properties in humans but was never marketed.

TPA-023 (MK-0777) is an anxiolytic drug with a novel chemical structure, which is used in scientific research. It has similar effects to benzodiazepine drugs, but is structurally distinct and so is classed as a nonbenzodiazepine anxiolytic. It is a mixed, subtype-selective ligand of the benzodiazepine site of α1, α2, α3, and α5-containing GABAA receptors, where it acts as a partial agonist at benzodiazepine sites of the α2 and α3-containing subtypes, but as a silent antagonist at α1 and α5-containing subtypes. It has primarily anxiolytic and anticonvulsant effects in animal tests, but with no sedative effects even at 50 times the effective anxiolytic dose.

Enobosarm, also formerly known as ostarine and by the developmental code names GTx-024, MK-2866, and S-22, is a selective androgen receptor modulator (SARM) which is under development for the treatment of androgen receptor-positive breast cancer in women and for improvement of body composition in people taking GLP-1 receptor agonists like semaglutide. It was also under development for a variety of other indications, including treatment of cachexia, Duchenne muscular dystrophy, muscle atrophy or sarcopenia, and stress urinary incontinence, but development for all other uses has been discontinued. Enobosarm was evaluated for the treatment of muscle wasting related to cancer in late-stage clinical trials, and the drug improved lean body mass in these trials, but it was not effective in improving muscle strength. As a result, enobosarm was not approved and development for this use was terminated. Enobosarm is taken by mouth.

Pomaglumetad (LY-404,039) is an amino acid analog drug that acts as a highly selective agonist for the metabotropic glutamate receptor group II subtypes mGluR2 and mGluR3. Pharmacological research has focused on its potential antipsychotic and anxiolytic effects. Pomaglumetad is intended as a treatment for schizophrenia and other psychotic and anxiety disorders by modulating glutamatergic activity and reducing presynaptic release of glutamate at synapses in limbic and forebrain areas relevant to these disorders. Human studies investigating therapeutic use of pomaglumetad have focused on the prodrug LY-2140023, a methionine amide of pomaglumetad (also called pomaglumetad methionil) since pomaglumetad exhibits low oral absorption and bioavailability in humans.
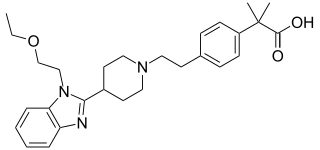
Bilastine is an antihistamine medication used to treat hives (urticaria), allergic rhinitis and itchy inflamed eyes (allergic conjunctivitis) caused by an allergy. It is a second-generation antihistamine and takes effect by selectively inhibiting the histamine H1 receptor, preventing these allergic reactions. Bilastine has an effectiveness similar to cetirizine, fexofenadine, and desloratadine.
