
Macrocephaly is a condition in which circumference of the human head is abnormally large. It may be pathological or harmless, and can be a familial genetic characteristic. People diagnosed with macrocephaly will receive further medical tests to determine whether the syndrome is accompanied by particular disorders. Those with benign or familial macrocephaly are considered to have megalencephaly.
Miller–Dieker syndrome, Miller–Dieker lissencephaly syndrome (MDLS), and chromosome 17p13.3 deletion syndrome is a micro deletion syndrome characterized by congenital malformations. Congenital malformations are physical defects detectable in an infant at birth which can involve many different parts of the body including the brain, hearts, lungs, liver, bones, or intestinal tract. MDS is a contiguous gene syndrome – a disorder due to the deletion of multiple gene loci adjacent to one another. The disorder arises from the deletion of part of the small arm of chromosome 17p, leading to partial monosomy. There may be unbalanced translocations, or the presence of a ring chromosome 17.

Cri du chat syndrome is a rare genetic disorder due to a partial chromosome deletion on chromosome 5. Its name is a French term referring to the characteristic cat-like cry of affected children. It was first described by Jérôme Lejeune in 1963. The condition affects an estimated 1 in 50,000 live births across all ethnicities and is more common in females by a 4:3 ratio.

A small supernumerary marker chromosome (sSMC) is an abnormal extra chromosome. It contains copies of parts of one or more normal chromosomes and like normal chromosomes is located in the cell's nucleus, is replicated and distributed into each daughter cell during cell division, and typically has genes which may be expressed. However, it may also be active in causing birth defects and neoplasms. The sSMC's small size makes it virtually undetectable using classical cytogenetic methods: the far larger DNA and gene content of the cell's normal chromosomes obscures those of the sSMC. Newer molecular techniques such as fluorescence in situ hybridization, next generation sequencing, comparative genomic hybridization, and highly specialized cytogenetic G banding analyses are required to study it. Using these methods, the DNA sequences and genes in sSMCs are identified and help define as well as explain any effect(s) it may have on individuals.

Saethre–Chotzen syndrome (SCS), also known as acrocephalosyndactyly type III, is a rare congenital disorder associated with craniosynostosis. This affects the shape of the head and face, resulting in a cone-shaped head and an asymmetrical face. Individuals with SCS also have droopy eyelids (ptosis), widely spaced eyes (hypertelorism), and minor abnormalities of the hands and feet (syndactyly). Individuals with more severe cases of SCS may have mild to moderate intellectual or learning disabilities. Depending on the level of severity, some individuals with SCS may require some form of medical or surgical intervention. Most individuals with SCS live fairly normal lives, regardless of whether medical treatment is needed or not.
Trisomy 8 causes Warkany syndrome 2, a human chromosomal disorder caused by having three copies (trisomy) of chromosome 8. It can appear with or without mosaicism.
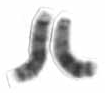
Chromosome 13 is one of the 23 pairs of chromosomes in humans. People normally have two copies of this chromosome. Chromosome 13 spans about 113 million base pairs and represents between 3.5 and 4% of the total DNA in cells.

GAPO syndrome is a rare, autosomal recessive disorder that causes severe growth retardation, and has been observed fewer than 30 times before 2011. GAPO is an acronym that encompasses the predominant traits of the disorder: growth retardation, alopecia, pseudoanodontia, and worsening optic atrophy in some subjects. Other common symptoms include premature aging, large, prominent foreheads, and delayed bone aging. GAPO syndrome typically results in premature death around age 30–40, due to interstitial fibrosis and atherosclerosis.

SCARF syndrome is a rare syndrome characterized by skeletal abnormalities, cutis laxa, craniostenosis, ambiguous genitalia, psychomotor retardation, and facial abnormalities. These characteristics are what make up the acronym SCARF. It shares some features with Lenz-Majewski hyperostotic dwarfism. It is a very rare disease with an incidence rate of approximately one in a million newborns. It has been clinically described in two males who were maternal cousins, as well as a 3-month-old female. Babies affected by this syndrome tend to have very loose skin, giving them an elderly facial appearance. Possible complications include dyspnea, abdominal hernia, heart disorders, joint disorders, and dislocations of multiple joints. It is believed that this disease's inheritance is X-linked recessive.

1p36 deletion syndrome is a congenital genetic disorder characterized by moderate to severe intellectual disability, delayed growth, hypotonia, seizures, limited speech ability, malformations, hearing and vision impairment, and distinct facial features. The symptoms may vary, depending on the exact location of the chromosomal deletion.

3C syndrome is a rare condition whose symptoms include heart defects, cerebellar hypoplasia, and cranial dysmorphism. It was first described in the medical literature in 1987 by Ritscher and Schinzel, for whom the disorder is sometimes named.

Young–Simpson syndrome (YSS) is a rare congenital disorder with symptoms including hypothyroidism, heart defects, facial dysmorphism, cryptorchidism in males, hypotonia, intellectual disability, and postnatal growth retardation.

Intermembrane lipid transfer protein VPS13B, also known as vacuolar protein sorting-associated 13B, and Cohen syndrome protein 1 is a protein that in humans is encoded by the VPS13B gene. It is a giant protein associated with the Golgi apparatus that is believed to be involved in post-Golgi apparatus sorting and trafficking. Mutations in the human VPS13B gene cause Cohen syndrome.
Smith–Fineman–Myers syndrome (SFMS1) is a congenital disorder that causes birth defects. This syndrome was named after Richard D. Smith, Robert M. Fineman and Gart G. Myers who discovered it around 1980.

Roberts syndrome, or sometimes called pseudothalidomide syndrome, is an extremely rare autosomal recessive genetic disorder that is characterized by mild to severe prenatal retardation or disruption of cell division, leading to malformation of the bones in the skull, face, arms, and legs.
Baller–Gerold syndrome (BGS) is a rare genetic syndrome that involves premature fusion of the skull bones and malformations of facial, forearm and hand bones. The symptoms of Baller–Gerold syndrome overlap with features of a few other genetics disorders: Rothmund–Thomson syndrome and RAPADILINO syndrome. The prevalence of BGS is unknown, as there have only been a few reported cases, but it is estimated to be less than 1 in a million. The name of the syndrome comes from the researchers Baller and Gerold who discovered the first three cases.
13q deletion syndrome is a rare genetic disease caused by the deletion of some or all of the large arm of human chromosome 13. Depending upon the size and location of the deletion on chromosome 13, the physical and mental manifestations will vary. It has the potential to cause intellectual disability and congenital malformations that affect a variety of organ systems. Because of the rarity of the disease in addition to the variations in the disease, the specific genes that cause this disease are unknown. This disease is also known as:
Fryns-Aftimos syndrome is a rare chromosomal condition and is associated with pachygyria, severe mental retardation, epilepsy and characteristic facial features. This syndrome is a malformation syndrome, characterized by numerous facial dysmorphias not limited to hypertelorism, iris or retinal coloboma, cleft lip, and congenital heart defects. This syndrome has been seen in 30 unrelated people. Characterized by a de novo mutation located on chromosome 7p22, there is typically no family history prior to onset. The severity of the disorder can be determined by the size of the deletion on 7p22, enveloping the ACTB gene and surrounding genes, which is consistent with a contiguous gene deletion syndrome. Confirming a diagnosis of Fryns-Aftimos syndrome typically consists of serial single-gene testing or multigene panel of genes of interest or exome sequencing.
Filippi syndrome, also known as Syndactyly Type I with Microcephaly and Mental Retardation, is a very rare autosomal recessive genetic disease. Only a very limited number of cases have been reported to date. Filippi Syndrome is associated with diverse symptoms of varying severity across affected individuals, for example malformation of digits, craniofacial abnormalities, intellectual disability, and growth retardation. The diagnosis of Filippi Syndrome can be done through clinical observation, radiography, and genetic testing. Filippi Syndrome cannot be cured directly as of 2022, hence the main focus of treatments is on tackling the symptoms observed on affected individuals. It was first reported in 1985.