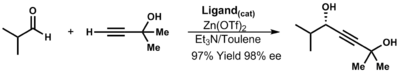Organometallic chemistry is the study of organometallic compounds, chemical compounds containing at least one chemical bond between a carbon atom of an organic molecule and a metal, including alkali, alkaline earth, and transition metals, and sometimes broadened to include metalloids like boron, silicon, and selenium, as well. Aside from bonds to organyl fragments or molecules, bonds to 'inorganic' carbon, like carbon monoxide, cyanide, or carbide, are generally considered to be organometallic as well. Some related compounds such as transition metal hydrides and metal phosphine complexes are often included in discussions of organometallic compounds, though strictly speaking, they are not necessarily organometallic. The related but distinct term "metalorganic compound" refers to metal-containing compounds lacking direct metal-carbon bonds but which contain organic ligands. Metal β-diketonates, alkoxides, dialkylamides, and metal phosphine complexes are representative members of this class. The field of organometallic chemistry combines aspects of traditional inorganic and organic chemistry.

Organolithium reagents are organometallic compounds that contain carbon–lithium bonds. These reagents are important in organic synthesis, and are frequently used to transfer the organic group or the lithium atom to the substrates in synthetic steps, through nucleophilic addition or simple deprotonation. Organolithium reagents are used in industry as an initiator for anionic polymerization, which leads to the production of various elastomers. They have also been applied in asymmetric synthesis in the pharmaceutical industry. Due to the large difference in electronegativity between the carbon atom and the lithium atom, the C−Li bond is highly ionic. Owing to the polar nature of the C−Li bond, organolithium reagents are good nucleophiles and strong bases. For laboratory organic synthesis, many organolithium reagents are commercially available in solution form. These reagents are highly reactive, and are sometimes pyrophoric.
The Stille reaction is a chemical reaction widely used in organic synthesis. The reaction involves the coupling of two organic groups, one of which is carried as an organotin compound. A variety of organic electrophiles provide the other coupling partner. The Stille reaction is one of many palladium-catalyzed coupling reactions.
The Suzuki reaction is an organic reaction, classified as a cross-coupling reaction, where the coupling partners are a boronic acid and an organohalide and the catalyst is a palladium(0) complex. It was first published in 1979 by Akira Suzuki, and he shared the 2010 Nobel Prize in Chemistry with Richard F. Heck and Ei-ichi Negishi for their contribution to the discovery and development of palladium-catalyzed cross-couplings in organic synthesis. This reaction is also known as the Suzuki–Miyaura reaction or simply as the Suzuki coupling. It is widely used to synthesize polyolefins, styrenes, and substituted biphenyls. Several reviews have been published describing advancements and the development of the Suzuki reaction. The general scheme for the Suzuki reaction is shown below, where a carbon-carbon single bond is formed by coupling a halide (R1-X) with an organoboron species (R2-BY2) using a palladium catalyst and a base.

The Simmons–Smith reaction is an organic cheletropic reaction involving an organozinc carbenoid that reacts with an alkene to form a cyclopropane. It is named after Howard Ensign Simmons, Jr. and Ronald D. Smith. It uses a methylene free radical intermediate that is delivered to both carbons of the alkene simultaneously, therefore the configuration of the double bond is preserved in the product and the reaction is stereospecific.
Metalation is a chemical reaction that forms a bond to a metal. This reaction usually refers to the replacement of a halogen atom in an organic molecule with a metal atom, resulting in an organometallic compound. In the laboratory, metalation is commonly used to activate organic molecules during the formation of C—X bonds, which are necessary for the synthesis of many organic molecules.

The Bamford–Stevens reaction is a chemical reaction whereby treatment of tosylhydrazones with strong base gives alkenes. It is named for the British chemist William Randall Bamford and the Scottish chemist Thomas Stevens Stevens (1900–2000). The usage of aprotic solvents gives predominantly Z-alkenes, while protic solvent gives a mixture of E- and Z-alkenes. As an alkene-generating transformation, the Bamford–Stevens reaction has broad utility in synthetic methodology and complex molecule synthesis.
The Hiyama coupling is a palladium-catalyzed cross-coupling reaction of organosilanes with organic halides used in organic chemistry to form carbon–carbon bonds. This reaction was discovered in 1988 by Tamejiro Hiyama and Yasuo Hatanaka as a method to form carbon-carbon bonds synthetically with chemo- and regioselectivity. The Hiyama coupling has been applied to the synthesis of various natural products.
The Corey–House synthesis is an organic reaction that involves the reaction of a lithium diorganylcuprate with an organic pseudohalide to form a new alkane, as well as an ill-defined organocopper species and lithium halide as byproducts.

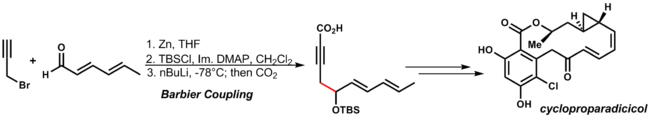
The Barbier reaction is an organometallic reaction between an alkyl halide, a carbonyl group and a metal. The reaction can be performed using magnesium, aluminium, zinc, indium, tin, samarium, barium or their salts. The reaction product is a primary, secondary or tertiary alcohol. The reaction is similar to the Grignard reaction but the crucial difference is that the organometallic species in the Barbier reaction is generated in situ, whereas a Grignard reagent is prepared separately before addition of the carbonyl compound. Unlike many Grignard reagents, the organometallic species generated in a Barbier reaction are unstable and thus cannot be stored or sold commercially. Barbier reactions are nucleophilic addition reactions that involve relatively inexpensive, water insensitive metals or metal compounds. For this reason it is possible in many cases to run the reaction in water, making the procedure part of green chemistry. In contrast, Grignard reagents and organolithium reagents are highly moisture sensitive and must be used under an inert atmosphere without the presence of water. The Barbier reaction is named after Victor Grignard's teacher Philippe Barbier.
The Reformatsky reaction is an organic reaction which condenses aldehydes or ketones with α-halo esters using metallic zinc to form β-hydroxy-esters:

A Grignard reagent or Grignard compound is a chemical compound with the generic formula R−Mg−X, where X is a halogen and R is an organic group, normally an alkyl or aryl. Two typical examples are methylmagnesium chloride Cl−Mg−CH3 and phenylmagnesium bromide (C6H5)−Mg−Br. They are a subclass of the organomagnesium compounds.

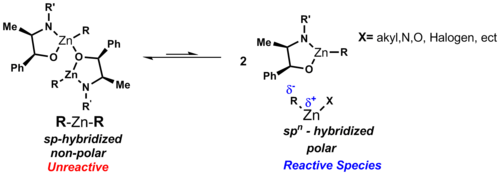
Diethylzinc (C2H5)2Zn, or DEZ, is a highly pyrophoric and reactive organozinc compound consisting of a zinc center bound to two ethyl groups. This colourless liquid is an important reagent in organic chemistry. It is available commercially as a solution in hexanes, heptane, or toluene, or as a pure liquid.
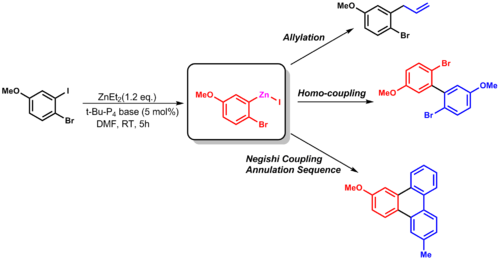
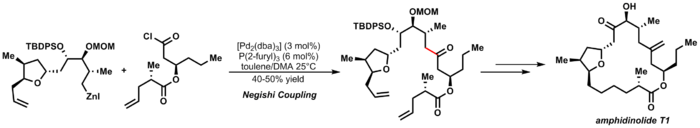
The Negishi coupling is a widely employed transition metal catalyzed cross-coupling reaction. The reaction couples organic halides or triflates with organozinc compounds, forming carbon-carbon bonds (C-C) in the process. A palladium (0) species is generally utilized as the metal catalyst, though nickel is sometimes used. A variety of nickel catalysts in either Ni0 or NiII oxidation state can be employed in Negishi cross couplings such as Ni(PPh3)4, Ni(acac)2, Ni(COD)2 etc.

A Rieke metal is a highly reactive metal powder generated by reduction of a metal salt with an alkali metal. These materials are named after Reuben D. Rieke, who first described the recipes for their preparation. Among the many metals that have been generated by this method are Mg, Ca, Ti, Fe, Co, Ni, Cu, Zn, and In, which in turn are called Rieke-magnesium, Rieke-calcium, etc.

Organocopper compounds is the chemistry of organometallic compounds containing a carbon to copper chemical bond. Organocopper chemistry is the study of organocopper compounds describing their physical properties, synthesis and reactions. They are reagents in organic chemistry.
In organic chemistry, the Kumada coupling is a type of cross coupling reaction, useful for generating carbon–carbon bonds by the reaction of a Grignard reagent and an organic halide. The procedure uses transition metal catalysts, typically nickel or palladium, to couple a combination of two alkyl, aryl or vinyl groups. The groups of Robert Corriu and Makoto Kumada reported the reaction independently in 1972.
Organomanganese chemistry is the chemistry of organometallic compounds containing a carbon to manganese chemical bond. In a 2009 review, Cahiez et al. argued that as manganese is cheap and benign, organomanganese compounds have potential as chemical reagents, although currently they are not widely used as such despite extensive research. A key disadvantage of organomanganese compounds is that they can be obtained directly from the metal only with difficulty.
Paul Knochel is a French chemist and a member of the French Academy of Sciences.
In organometallic chemistry, metal–halogen exchange is a fundamental reaction that converts a organic halide into an organometallic product. The reaction commonly involves the use of electropositive metals and organochlorides, bromides, and iodides. Particularly well-developed is the use of metal–halogen exchange for the preparation of organolithium compounds.