Elias James Corey is an American organic chemist. In 1990, he won the Nobel Prize in Chemistry "for his development of the theory and methodology of organic synthesis", specifically retrosynthetic analysis. Regarded by many as one of the greatest living chemists, he has developed numerous synthetic reagents, methodologies and total syntheses and has advanced the science of organic synthesis considerably.

The Sharpless epoxidation reaction is an enantioselective chemical reaction to prepare 2,3-epoxyalcohols from primary and secondary allylic alcohols. The oxidizing agent is tert-butyl hydroperoxide. The method relies on a catalyst formed from titanium tetra(isopropoxide) and diethyl tartrate.
An ylide or ylid is a neutral dipolar molecule containing a formally negatively charged atom (usually a carbanion) directly attached to a heteroatom with a formal positive charge (usually nitrogen, phosphorus or sulfur), and in which both atoms have full octets of electrons. The result can be viewed as a structure in which two adjacent atoms are connected by both a covalent and an ionic bond; normally written X+–Y−. Ylides are thus 1,2-dipolar compounds, and a subclass of zwitterions. They appear in organic chemistry as reagents or reactive intermediates.

In organometallic chemistry, organolithium reagents are chemical compounds that contain carbon–lithium (C–Li) bonds. These reagents are important in organic synthesis, and are frequently used to transfer the organic group or the lithium atom to the substrates in synthetic steps, through nucleophilic addition or simple deprotonation. Organolithium reagents are used in industry as an initiator for anionic polymerization, which leads to the production of various elastomers. They have also been applied in asymmetric synthesis in the pharmaceutical industry. Due to the large difference in electronegativity between the carbon atom and the lithium atom, the C−Li bond is highly ionic. Owing to the polar nature of the C−Li bond, organolithium reagents are good nucleophiles and strong bases. For laboratory organic synthesis, many organolithium reagents are commercially available in solution form. These reagents are highly reactive, and are sometimes pyrophoric.

In organic chemistry, the ene reaction is a chemical reaction between an alkene with an allylic hydrogen and a compound containing a multiple bond, in order to form a new σ-bond with migration of the ene double bond and 1,5 hydrogen shift. The product is a substituted alkene with the double bond shifted to the allylic position.
The Simmons–Smith reaction is an organic cheletropic reaction involving an organozinc carbenoid that reacts with an alkene to form a cyclopropane. It is named after Howard Ensign Simmons, Jr. and Ronald D. Smith. It uses a methylene free radical intermediate that is delivered to both carbons of the alkene simultaneously, therefore the configuration of the double bond is preserved in the product and the reaction is stereospecific.
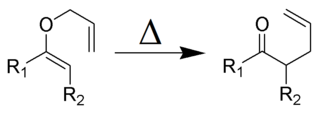
The Claisen rearrangement is a powerful carbon–carbon bond-forming chemical reaction discovered by Rainer Ludwig Claisen. The heating of an allyl vinyl ether will initiate a [3,3]-sigmatropic rearrangement to give a γ,δ-unsaturated carbonyl, driven by exergonically favored carbonyl CO bond formation.
The Carroll rearrangement is a rearrangement reaction in organic chemistry and involves the transformation of a β-keto allyl ester into a α-allyl-β-ketocarboxylic acid. This organic reaction is accompanied by decarboxylation and the final product is a γ,δ-allylketone. The Carroll rearrangement is an adaptation of the Claisen rearrangement and effectively a decarboxylative allylation.
The Overman rearrangement is a chemical reaction that can be described as a Claisen rearrangement of allylic alcohols to give allylic trichloroacetamides through an imidate intermediate. The Overman rearrangement was discovered in 1974 by Larry Overman.

In stereochemistry, a chiral auxiliary is a stereogenic group or unit that is temporarily incorporated into an organic compound in order to control the stereochemical outcome of the synthesis. The chirality present in the auxiliary can bias the stereoselectivity of one or more subsequent reactions. The auxiliary can then be typically recovered for future use.

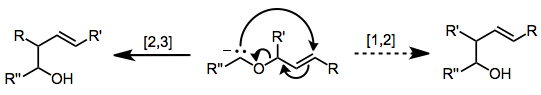
2,3-Sigmatropic rearrangements are a type of sigmatropic rearrangements and can be classified into two types. Rearrangements of allylic sulfoxides, amine oxides, selenoxides are neutral. Rearrangements of carbanions of allyl ethers are anionic. The general scheme for this kind of rearrangement is:
The Nazarov cyclization reaction is a chemical reaction used in organic chemistry for the synthesis of cyclopentenones. The reaction is typically divided into classical and modern variants, depending on the reagents and substrates employed. It was originally discovered by Ivan Nikolaevich Nazarov (1906–1957) in 1941 while studying the rearrangements of allyl vinyl ketones.
The Kornblum–DeLaMare rearrangement is a rearrangement reaction in organic chemistry in which a primary or secondary organic peroxide is converted to the corresponding ketone and alcohol under acid or base catalysis. The reaction is relevant as a tool in organic synthesis and is a key step in the biosynthesis of prostaglandins.
The Rubottom oxidation is a useful, high-yielding chemical reaction between silyl enol ethers and peroxyacids to give the corresponding α-hydroxy carbonyl product. The mechanism of the reaction was proposed in its original disclosure by A.G. Brook with further evidence later supplied by George M. Rubottom. After a Prilezhaev-type oxidation of the silyl enol ether with the peroxyacid to form the siloxy oxirane intermediate, acid-catalyzed ring-opening yields an oxocarbenium ion. This intermediate then participates in a 1,4-silyl migration to give an α-siloxy carbonyl derivative that can be readily converted to the α-hydroxy carbonyl compound in the presence of acid, base, or a fluoride source.

Allylic strain in organic chemistry is a type of strain energy resulting from the interaction between a substituent on one end of an olefin with an allylic substituent on the other end. If the substituents are large enough in size, they can sterically interfere with each other such that one conformer is greatly favored over the other. Allylic strain was first recognized in the literature in 1965 by Johnson and Malhotra. The authors were investigating cyclohexane conformations including endocyclic and exocylic double bonds when they noticed certain conformations were disfavored due to the geometry constraints caused by the double bond. Organic chemists capitalize on the rigidity resulting from allylic strain for use in asymmetric reactions.
Organostannane addition reactions comprise the nucleophilic addition of an allyl-, allenyl-, or propargylstannane to an aldehyde, imine, or, in rare cases, a ketone. The reaction is widely used for carbonyl allylation.
Electrophilic amination is a chemical process involving the formation of a carbon–nitrogen bond through the reaction of a nucleophilic carbanion with an electrophilic source of nitrogen.
The Tsuji–Trost reaction is a palladium-catalysed substitution reaction involving a substrate that contains a leaving group in an allylic position. The palladium catalyst first coordinates with the allyl group and then undergoes oxidative addition, forming the π-allyl complex. This allyl complex can then be attacked by a nucleophile, resulting in the substituted product.
William Clark Still is an American organic chemist. As a distinguished professor at Columbia University, Clark Still made significant contributions to the field of organic chemistry, particularly in the areas of natural product synthesis, reaction development, conformational analysis, macrocyclic stereocontrol, and computational chemistry. Still and coworkers also developed the purification technique known as flash column chromatography which is widely used for the purification of organic compounds.
In organic chemistry, the oxy-Cope rearrangement is a chemical reaction. It involves reorganization of the skeleton of certain unsaturated alcohols. It is a variation of the Cope rearrangement in which 1,5-dien-3-ols are converted to unsaturated carbonyl compounds by a mechanism typical for such a [3,3]-sigmatropic rearrangement.