The Beckmann rearrangement, named after the German chemist Ernst Otto Beckmann (1853–1923), is a rearrangement of an oxime functional group to substituted amides. The rearrangement has also been successfully performed on haloimines and nitrones. Cyclic oximes and haloimines yield lactams.
An ylide or ylid is a neutral dipolar molecule containing a formally negatively charged atom (usually a carbanion) directly attached to a heteroatom with a formal positive charge (usually nitrogen, phosphorus or sulfur), and in which both atoms have full octets of electrons. The result can be viewed as a structure in which two adjacent atoms are connected by both a covalent and an ionic bond; normally written X+–Y−. Ylides are thus 1,2-dipolar compounds, and a subclass of zwitterions. They appear in organic chemistry as reagents or reactive intermediates.

In organic chemistry, an imine is a functional group or organic compound containing a carbon–nitrogen double bond. The nitrogen atom can be attached to a hydrogen or an organic group (R). The carbon atom has two additional single bonds. Imines are common in synthetic and naturally occurring compounds and they participate in many reactions.

Oxazole is the parent compound for a vast class of heterocyclic aromatic organic compounds. These are azoles with an oxygen and a nitrogen separated by one carbon. Oxazoles are aromatic compounds but less so than the thiazoles. Oxazole is a weak base; its conjugate acid has a pKa of 0.8, compared to 7 for imidazole.
Isoxazole is an electron-rich azole with an oxygen atom next to the nitrogen. It is also the class of compounds containing this ring. Isoxazolyl is the univalent functional group derived from isoxazole.
The Passerini reaction is a chemical reaction involving an isocyanide, an aldehyde, and a carboxylic acid to form a α-acyloxy amide. This addition reaction is one of the oldest isocyanide-based multicomponent reactions and was first described in 1921 by Mario Passerini in Florence, Italy. It is typically carried out in aprotic solvents but can also be performed in ionic liquids such as water or deep eutectic solvents. It is a third order reaction; first order in each of the reactants. The Passerini reaction is often used in combinatorial and medicinal chemistry with recent utility in green chemistry and polymer chemistry. As isocyanides exhibit high functional group tolerance, chemoselectivity, regioselectivity, and stereoselectivity, the Passerini reaction has a wide range of synthetic applications.
The Fischer oxazole synthesis is a chemical synthesis of an oxazole from a cyanohydrin and an aldehyde in the presence of anhydrous hydrochloric acid. This method was discovered by Emil Fischer in 1896. The cyanohydrin itself is derived from a separate aldehyde. The reactants of the oxazole synthesis itself, the cyanohydrin of an aldehyde and the other aldehyde itself, are usually present in equimolar amounts. Both reactants usually have an aromatic group, which appear at specific positions on the resulting heterocycle.

The Claisen rearrangement is a powerful carbon–carbon bond-forming chemical reaction discovered by Rainer Ludwig Claisen. The heating of an allyl vinyl ether will initiate a [3,3]-sigmatropic rearrangement to give a γ,δ-unsaturated carbonyl, driven by exergonically favored carbonyl CO bond formation (Δ = −327 kcal/mol.
The Ullmann reaction or Ullmann coupling, named after Fritz Ullmann, couples two aryl or alkyl groups with the help of copper. The reaction was first reported by Ullmann and his student Bielecki in 1901. It has been later shown that palladium and nickel can also be effectively used.
The Baeyer–Villiger oxidation is an organic reaction that forms an ester from a ketone or a lactone from a cyclic ketone, using peroxyacids or peroxides as the oxidant. The reaction is named after Adolf von Baeyer and Victor Villiger who first reported the reaction in 1899.

Prismane or 'Ladenburg benzene' is a polycyclic hydrocarbon with the formula C6H6. It is an isomer of benzene, specifically a valence isomer. Prismane is far less stable than benzene. The carbon (and hydrogen) atoms of the prismane molecule are arranged in the shape of a six-atom triangular prism—this compound is the parent and simplest member of the prismanes class of molecules. Albert Ladenburg proposed this structure for the compound now known as benzene. The compound was not synthesized until 1973.
In organic chemistry, a cyclophane is a hydrocarbon consisting of an aromatic unit and a chain that forms a bridge between two non-adjacent positions of the aromatic ring. More complex derivatives with multiple aromatic units and bridges forming cagelike structures are also known. Cyclophanes are well-studied examples of strained organic compounds.

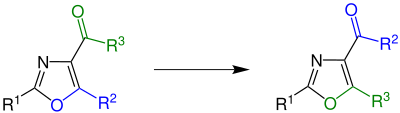
The Robinson–Gabriel synthesis is an organic reaction in which a 2-acylamino-ketone reacts intramolecularly followed by a dehydration to give an oxazole. A cyclodehydrating agent is needed to catalyze the reaction It is named after Sir Robert Robinson and Siegmund Gabriel who described the reaction in 1909 and 1910, respectively.

The Bartoli indole synthesis is the chemical reaction of ortho-substituted nitroarenes and nitrosoarenes with vinyl Grignard reagents to form substituted indoles.
In organic chemistry, the Paal–Knorr synthesis is a reaction used to synthesize substituted furans, pyrroles, or thiophenes from 1,4-diketones. It is a synthetically valuable method for obtaining substituted furans and pyrroles, which are common structural components of many natural products. It was initially reported independently by German chemists Carl Paal and Ludwig Knorr in 1884 as a method for the preparation of furans, and has been adapted for pyrroles and thiophenes. Although the Paal–Knorr synthesis has seen widespread use, the mechanism wasn't fully understood until it was elucidated by V. Amarnath et al. in the 1990s.
The Kulinkovich reaction describes the organic synthesis of substituted cyclopropanols through reaction of esters with dialkyldialkoxytitanium reagents, which are generated in situ from Grignard reagents containing a hydrogen in beta-position and titanium(IV) alkoxides such as titanium isopropoxide. This reaction was first reported by Oleg Kulinkovich and coworkers in 1989.
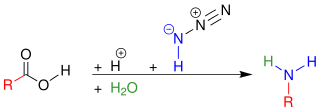
In organic chemistry, the Schmidt reaction is an organic reaction in which an azide reacts with a carbonyl derivative, usually an aldehyde, ketone, or carboxylic acid, under acidic conditions to give an amine or amide, with expulsion of nitrogen. It is named after Karl Friedrich Schmidt (1887–1971), who first reported it in 1924 by successfully converting benzophenone and hydrazoic acid to benzanilide. The intramolecular reaction was not reported until 1991 but has become important in the synthesis of natural products. The reaction is effective with carboxylic acids to give amines (above), and with ketones to give amides (below).
The Stieglitz rearrangement is a rearrangement reaction in organic chemistry which is named after the American chemist Julius Stieglitz (1867–1937) and was first investigated by him and Paul Nicholas Leech in 1913. It describes the 1,2-rearrangement of trityl amine derivatives to triaryl imines. It is comparable to a Beckmann rearrangement which also involves a substitution at a nitrogen atom through a carbon to nitrogen shift. As an example, triaryl hydroxylamines can undergo a Stieglitz rearrangement by dehydration and the shift of a phenyl group after activation with phosphorus pentachloride to yield the respective triaryl imine, a Schiff base.
The vinylcyclopropane rearrangement or vinylcyclopropane-cyclopentene rearrangement is a ring expansion reaction, converting a vinyl-substituted cyclopropane ring into a cyclopentene ring.
An insertion reaction is a chemical reaction where one chemical entity interposes itself into an existing bond of typically a second chemical entity e.g.: