In organometallic chemistry, organolithium reagents are chemical compounds that contain carbon–lithium (C–Li) bonds. These reagents are important in organic synthesis, and are frequently used to transfer the organic group or the lithium atom to the substrates in synthetic steps, through nucleophilic addition or simple deprotonation. Organolithium reagents are used in industry as an initiator for anionic polymerization, which leads to the production of various elastomers. They have also been applied in asymmetric synthesis in the pharmaceutical industry. Due to the large difference in electronegativity between the carbon atom and the lithium atom, the C−Li bond is highly ionic. Owing to the polar nature of the C−Li bond, organolithium reagents are good nucleophiles and strong bases. For laboratory organic synthesis, many organolithium reagents are commercially available in solution form. These reagents are highly reactive, and are sometimes pyrophoric.
In organic chemistry, an electrocyclic reaction is a type of pericyclic rearrangement where the net result is one pi bond being converted into one sigma bond or vice versa. These reactions are usually categorized by the following criteria:
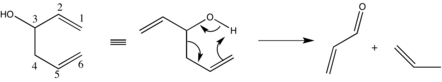
A sigmatropic reaction in organic chemistry is a pericyclic reaction wherein the net result is one σ-bond is changed to another σ-bond in an uncatalyzed intramolecular reaction. The name sigmatropic is the result of a compounding of the long-established sigma designation from single carbon–carbon bonds and the Greek word tropos, meaning turn. In this type of rearrangement reaction, a substituent moves from one part of a π-bonded system to another part in an intramolecular reaction with simultaneous rearrangement of the π system. True sigmatropic reactions are usually uncatalyzed, although Lewis acid catalysis is possible. Sigmatropic reactions often have transition-metal catalysts that form intermediates in analogous reactions. The most well-known of the sigmatropic rearrangements are the [3,3] Cope rearrangement, Claisen rearrangement, Carroll rearrangement, and the Fischer indole synthesis.
The Cope rearrangement is an extensively studied organic reaction involving the [3,3]-sigmatropic rearrangement of 1,5-dienes. It was developed by Arthur C. Cope and Elizabeth Hardy. For example, 3-methyl-hexa-1,5-diene heated to 300 °C yields hepta-1,5-diene.
Organopalladium chemistry is a branch of organometallic chemistry that deals with organic palladium compounds and their reactions. Palladium is often used as a catalyst in the reduction of alkenes and alkynes with hydrogen. This process involves the formation of a palladium-carbon covalent bond. Palladium is also prominent in carbon-carbon coupling reactions, as demonstrated in tandem reactions.

The Bamford–Stevens reaction is a chemical reaction whereby treatment of tosylhydrazones with strong base gives alkenes. It is named for the British chemist William Randall Bamford and the Scottish chemist Thomas Stevens Stevens (1900–2000). The usage of aprotic solvents gives predominantly Z-alkenes, while protic solvent gives a mixture of E- and Z-alkenes. As an alkene-generating transformation, the Bamford–Stevens reaction has broad utility in synthetic methodology and complex molecule synthesis.
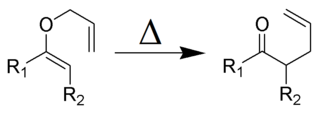
The Claisen rearrangement is a powerful carbon–carbon bond-forming chemical reaction discovered by Rainer Ludwig Claisen. The heating of an allyl vinyl ether will initiate a [3,3]-sigmatropic rearrangement to give a γ,δ-unsaturated carbonyl, driven by exergonically favored carbonyl CO bond formation.
The Overman rearrangement is a chemical reaction that can be described as a Claisen rearrangement of allylic alcohols to give allylic trichloroacetamides through an imidate intermediate. The Overman rearrangement was discovered in 1974 by Larry Overman.
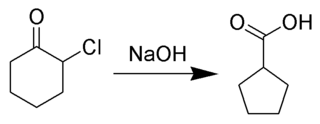
The Favorskii rearrangement is principally a rearrangement of cyclopropanones and α-halo ketones that leads to carboxylic acid derivatives. In the case of cyclic α-halo ketones, the Favorskii rearrangement constitutes a ring contraction. This rearrangement takes place in the presence of a base, sometimes hydroxide, to yield a carboxylic acid but most of the time either an alkoxide base or an amine to yield an ester or an amide, respectively. α,α'-Dihaloketones eliminate HX under the reaction conditions to give α,β-unsaturated carbonyl compounds.

The Baker–Venkataraman rearrangement is the chemical reaction of 2-acetoxyacetophenones with base to form 1,3-diketones.
Bullvalene is a hydrocarbon with the chemical formula C10H10. The molecule has a cage-like structure formed by the fusion of one cyclopropane and three cyclohepta-1,4-diene rings. Bullvalene is unusual as an organic molecule due to the C−C and C=C bonds forming and breaking rapidly on the NMR timescale; this property makes it a fluxional molecule.

The Wolff rearrangement is a reaction in organic chemistry in which an α-diazocarbonyl compound is converted into a ketene by loss of dinitrogen with accompanying 1,2-rearrangement. The Wolff rearrangement yields a ketene as an intermediate product, which can undergo nucleophilic attack with weakly acidic nucleophiles such as water, alcohols, and amines, to generate carboxylic acid derivatives or undergo [2+2] cycloaddition reactions to form four-membered rings. The mechanism of the Wolff rearrangement has been the subject of debate since its first use. No single mechanism sufficiently describes the reaction, and there are often competing concerted and carbene-mediated pathways; for simplicity, only the textbook, concerted mechanism is shown below. The reaction was discovered by Ludwig Wolff in 1902. The Wolff rearrangement has great synthetic utility due to the accessibility of α-diazocarbonyl compounds, variety of reactions from the ketene intermediate, and stereochemical retention of the migrating group. However, the Wolff rearrangement has limitations due to the highly reactive nature of α-diazocarbonyl compounds, which can undergo a variety of competing reactions.
The Kornblum–DeLaMare rearrangement is a rearrangement reaction in organic chemistry in which a primary or secondary organic peroxide is converted to the corresponding ketone and alcohol under acid or base catalysis. The reaction is relevant as a tool in organic synthesis and is a key step in the biosynthesis of prostaglandins.
The Rubottom oxidation is a useful, high-yielding chemical reaction between silyl enol ethers and peroxyacids to give the corresponding α-hydroxy carbonyl product. The mechanism of the reaction was proposed in its original disclosure by A.G. Brook with further evidence later supplied by George M. Rubottom. After a Prilezhaev-type oxidation of the silyl enol ether with the peroxyacid to form the siloxy oxirane intermediate, acid-catalyzed ring-opening yields an oxocarbenium ion. This intermediate then participates in a 1,4-silyl migration to give an α-siloxy carbonyl derivative that can be readily converted to the α-hydroxy carbonyl compound in the presence of acid, base, or a fluoride source.
The Ireland–Claisen rearrangement is a chemical reaction of an allylic ester with strong base to give an γ,δ-unsaturated carboxylic acid.
The vinylcyclopropane rearrangement or vinylcyclopropane-cyclopentene rearrangement is a ring expansion reaction, converting a vinyl-substituted cyclopropane ring into a cyclopentene ring.
Electrophilic amination is a chemical process involving the formation of a carbon–nitrogen bond through the reaction of a nucleophilic carbanion with an electrophilic source of nitrogen.
The [2,3]-Wittig rearrangement is the transformation of an allylic ether into a homoallylic alcohol via a concerted, pericyclic process. Because the reaction is concerted, it exhibits a high degree of stereocontrol, and can be employed early in a synthetic route to establish stereochemistry. The Wittig rearrangement requires strongly basic conditions, however, as a carbanion intermediate is essential. [1,2]-Wittig rearrangement is a competitive process.
William Clark Still is an American organic chemist. As a distinguished professor at Columbia University, Clark Still made significant contributions to the field of organic chemistry, particularly in the areas of natural product synthesis, reaction development, conformational analysis, macrocyclic stereocontrol, and computational chemistry. Still and coworkers also developed the purification technique known as flash column chromatography which is widely used for the purification of organic compounds.
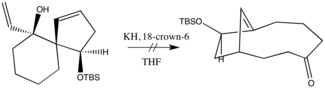
Rearrangements, especially those that can participate in cascade reactions, such as the aza-Cope rearrangements, are of high practical as well as conceptual importance in organic chemistry, due to their ability to quickly build structural complexity out of simple starting materials. The aza-Cope rearrangements are examples of heteroatom versions of the Cope rearrangement, which is a [3,3]-sigmatropic rearrangement that shifts single and double bonds between two allylic components. In accordance with the Woodward-Hoffman rules, thermal aza-Cope rearrangements proceed suprafacially. Aza-Cope rearrangements are generally classified by the position of the nitrogen in the molecule :