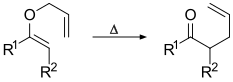An ylide or ylid is a neutral dipolar molecule containing a formally negatively charged atom (usually a carbanion) directly attached to a heteroatom with a formal positive charge (usually nitrogen, phosphorus or sulfur), and in which both atoms have full octets of electrons. The result can be viewed as a structure in which two adjacent atoms are connected by both a covalent and an ionic bond; normally written X+–Y−. Ylides are thus 1,2-dipolar compounds, and a subclass of zwitterions. They appear in organic chemistry as reagents or reactive intermediates.

In organometallic chemistry, organolithium reagents are chemical compounds that contain carbon–lithium (C–Li) bonds. These reagents are important in organic synthesis, and are frequently used to transfer the organic group or the lithium atom to the substrates in synthetic steps, through nucleophilic addition or simple deprotonation. Organolithium reagents are used in industry as an initiator for anionic polymerization, which leads to the production of various elastomers. They have also been applied in asymmetric synthesis in the pharmaceutical industry. Due to the large difference in electronegativity between the carbon atom and the lithium atom, the C−Li bond is highly ionic. Owing to the polar nature of the C−Li bond, organolithium reagents are good nucleophiles and strong bases. For laboratory organic synthesis, many organolithium reagents are commercially available in solution form. These reagents are highly reactive, and are sometimes pyrophoric.
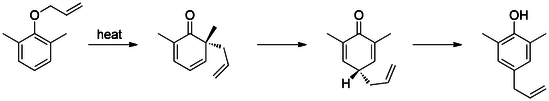
A sigmatropic reaction in organic chemistry is a pericyclic reaction wherein the net result is one σ-bond is changed to another σ-bond in an uncatalyzed intramolecular reaction. The name sigmatropic is the result of a compounding of the long-established sigma designation from single carbon–carbon bonds and the Greek word tropos, meaning turn. In this type of rearrangement reaction, a substituent moves from one part of a π-bonded system to another part in an intramolecular reaction with simultaneous rearrangement of the π system. True sigmatropic reactions are usually uncatalyzed, although Lewis acid catalysis is possible. Sigmatropic reactions often have transition-metal catalysts that form intermediates in analogous reactions. The most well-known of the sigmatropic rearrangements are the [3,3] Cope rearrangement, Claisen rearrangement, Carroll rearrangement, and the Fischer indole synthesis.

In organic chemistry, the Michael reaction or Michael 1,4 addition is a reaction between a Michael donor and a Michael acceptor to produce a Michael adduct by creating a carbon-carbon bond at the acceptor's β-carbon. It belongs to the larger class of conjugate additions and is widely used for the mild formation of carbon-carbon bonds.

In organic chemistry, enolates are organic anions derived from the deprotonation of carbonyl compounds. Rarely isolated, they are widely used as reagents in the synthesis of organic compounds.
The Simmons–Smith reaction is an organic cheletropic reaction involving an organozinc carbenoid that reacts with an alkene to form a cyclopropane. It is named after Howard Ensign Simmons, Jr. and Ronald D. Smith. It uses a methylene free radical intermediate that is delivered to both carbons of the alkene simultaneously, therefore the configuration of the double bond is preserved in the product and the reaction is stereospecific.
The Carroll rearrangement is a rearrangement reaction in organic chemistry and involves the transformation of a β-keto allyl ester into a α-allyl-β-ketocarboxylic acid. This organic reaction is accompanied by decarboxylation and the final product is a γ,δ-allylketone. The Carroll rearrangement is an adaptation of the Claisen rearrangement and effectively a decarboxylative allylation.
The Overman rearrangement is a chemical reaction that can be described as a Claisen rearrangement of allylic alcohols to give allylic trichloroacetamides through an imidate intermediate. The Overman rearrangement was discovered in 1974 by Larry Overman.

In stereochemistry, a chiral auxiliary is a stereogenic group or unit that is temporarily incorporated into an organic compound in order to control the stereochemical outcome of the synthesis. The chirality present in the auxiliary can bias the stereoselectivity of one or more subsequent reactions. The auxiliary can then be typically recovered for future use.

The Baker–Venkataraman rearrangement is the chemical reaction of 2-acetoxyacetophenones with base to form 1,3-diketones.

In organic chemistry, an ortho ester is a functional group containing three alkoxy groups attached to one carbon atom, i.e. with the general formula RC(OR′)3. Orthoesters may be considered as products of exhaustive alkylation of unstable orthocarboxylic acids and it is from these that the name 'ortho ester' is derived. An example is ethyl orthoacetate, CH3C(OCH2CH3)3, more correctly known as 1,1,1-triethoxyethane.
The Ramberg–Bäcklund reaction is an organic reaction converting an α-halo sulfone into an alkene in presence of a base with extrusion of sulfur dioxide. The reaction is named after the two Swedish chemists Ludwig Ramberg and Birger Bäcklund. The carbanion formed by deprotonation gives an unstable episulfone that decomposes with elimination of sulfur dioxide. This elimination step is considered to be a concerted cycloelimination.
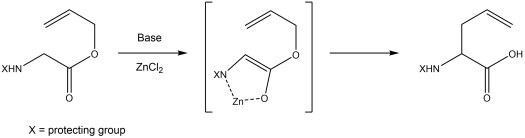
The Ireland–Claisen rearrangement is a chemical reaction of an allylic ester with strong base to give an γ,δ-unsaturated carboxylic acid.

Larry E. Overman is Distinguished Professor of Chemistry at the University of California, Irvine. He was born in Chicago in 1943. Overman obtained a B.A. degree from Earlham College in 1965, and he completed his Ph.D. in chemistry from the University of Wisconsin–Madison in 1969, under Howard Whitlock Jr. Professor Overman is a member of the United States National Academy of Sciences and the American Academy of Arts and Sciences. He was the recipient of the Arthur C. Cope Award in 2003, and he was awarded the Tetrahedron Prize for Creativity in Organic Chemistry for 2008.
The [2,3]-Wittig rearrangement is the transformation of an allylic ether into a homoallylic alcohol via a concerted, pericyclic process. Because the reaction is concerted, it exhibits a high degree of stereocontrol, and can be employed early in a synthetic route to establish stereochemistry. The Wittig rearrangement requires strongly basic conditions, however, as a carbanion intermediate is essential. [1,2]-Wittig rearrangement is a competitive process.
William Clark Still is an American organic chemist. As a distinguished professor at Columbia University, Clark Still made significant contributions to the field of organic chemistry, particularly in the areas of natural product synthesis, reaction development, conformational analysis, macrocyclic stereocontrol, and computational chemistry. Still and coworkers also developed the purification technique known as flash column chromatography which is widely used for the purification of organic compounds.
Rearrangements, especially those that can participate in cascade reactions, such as the aza-Cope rearrangements, are of high practical as well as conceptual importance in organic chemistry, due to their ability to quickly build structural complexity out of simple starting materials. The aza-Cope rearrangements are examples of heteroatom versions of the Cope rearrangement, which is a [3,3]-sigmatropic rearrangement that shifts single and double bonds between two allylic components. In accordance with the Woodward-Hoffman rules, thermal aza-Cope rearrangements proceed suprafacially. Aza-Cope rearrangements are generally classified by the position of the nitrogen in the molecule :
Modified Wittig–Claisen tandem reaction is a cascade reaction that combines the Wittig reaction and Claisen rearrangement together. The Wittig reaction generates the allyl vinyl ether intermediate that further participates in a Claisen rearrangement to generate the final γ,δ-unsaturated ketone or aldehyde product.
In organic chemistry, the oxy-Cope rearrangement is a chemical reaction. It involves reorganization of the skeleton of certain unsaturated alcohols. It is a variation of the Cope rearrangement in which 1,5-dien-3-ols are converted to unsaturated carbonyl compounds by a mechanism typical for such a [3,3]-sigmatropic rearrangement.