Kabuki syndrome is a pediatric congenital disorder of genetic origin. It affects multiple parts of the body with varying symptoms and severity, although the most common is the characteristic facial appearance. It is quite rare, affecting roughly one in 32,000 births. It was first identified and described in 1981 by two Japanese groups, led by scientists Norio Niikawa and Yoshikazu Kuroki. It is named Kabuki syndrome because of the facial resemblance of affected individuals to stage makeup used in kabuki, a Japanese traditional theatrical form.
CHARGE syndrome is a rare syndrome caused by a genetic disorder. First described in 1979, the acronym "CHARGE" came into use for newborn children with the congenital features of coloboma of the eye, heart defects, atresia of the nasal choanae, retardation of growth and/or development, genital and/or urinary abnormalities, and ear abnormalities and deafness. These features are no longer used in making a diagnosis of CHARGE syndrome, but the name remains. About two thirds of cases are due to a CHD7 mutation. CHARGE syndrome occurs only in 0.1–1.2 per 10,000 live births; as of 2009 it was the leading cause of congenital deafblindness in the US.

Fraser syndrome is an autosomal recessive congenital disorder. Fraser syndrome is named for the geneticist George R. Fraser, who first described the syndrome in 1962.

Simpson–Golabi–Behmel syndrome (SGBS), is a rare inherited congenital disorder that can cause craniofacial, skeletal, cardiac, and renal abnormalities. The syndrome is inherited in an X-linked recessive fashion, where males express the phenotype and females usually do not. Females that possess one copy of the mutation are considered to be carriers of the syndrome and may express varying degrees of the phenotype.

22q13 deletion syndrome, also known as Phelan–McDermid syndrome (PMS), is a genetic disorder caused by deletions or rearrangements on the q terminal end of chromosome 22. Any abnormal genetic variation in the q13 region that presents with significant manifestations (phenotype) typical of a terminal deletion may be diagnosed as 22q13 deletion syndrome. There is disagreement among researchers as to the exact definition of 22q13 deletion syndrome. The Developmental Synaptopathies Consortium defines PMS as being caused by SHANK3 mutations, a definition that appears to exclude terminal deletions. The requirement to include SHANK3 in the definition is supported by many but not by those who first described 22q13 deletion syndrome.

A chromosomal disorder, anomaly, aberration, or mutation is a missing, extra, or irregular portion of chromosomal DNA. It can be from a typical number of chromosomes or a structural abnormality in one or more chromosomes. Chromosome mutation was formerly used in a strict sense to mean a change in a chromosomal segment, involving more than one gene. The term "karyotype" refers to the full set of chromosomes from an individual; this can be compared to a "normal" karyotype for the species via genetic testing. A chromosome anomaly may be detected or confirmed in this manner. Chromosome anomalies usually occur when there is an error in cell division following meiosis or mitosis. There are many types of chromosome anomalies. They can be organized into two basic groups, numerical and structural anomalies.

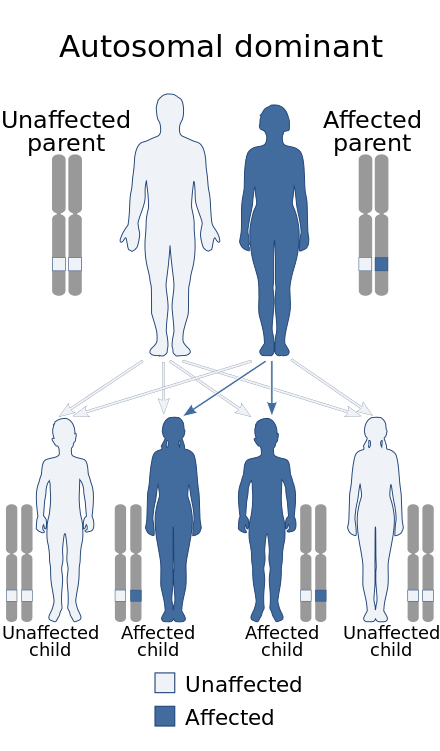
Papillorenal syndrome, is an autosomal dominant genetic disorder marked by underdevelopment (hypoplasia) of the kidney and colobomas of the optic nerve.

Frank ter Haar-syndrome (FTHS), also known as Ter Haar-syndrome, is a rare disease characterized by abnormalities that affect bone, heart, and eye development. Children born with the disease usually die very young.

Angelman syndrome (AS) is a genetic disorder that mainly affects the nervous system. Symptoms include a small head and a specific facial appearance, severe intellectual disability, developmental disability, speaking problems, balance and movement problems, seizures, and sleep problems. Children usually have a happy personality and have a particular interest in water. The symptoms generally become noticeable by one year of age.
Congenital generalized lipodystrophy is an extremely rare autosomal recessive condition, characterized by an extreme scarcity of fat in the subcutaneous tissues. It is a type of lipodystophy disorder where the magnitude of fat loss determines the severity of metabolic complications. Only 250 cases of the condition have been reported, and it is estimated that it occurs in 1 in 10 million people worldwide.
A Finnish heritage disease is a genetic disease or disorder that is significantly more common in people whose ancestors were ethnic Finns, natives of Finland and Sweden (Meänmaa) and Russia. 36 rare diseases are regarded as Finnish heritage diseases. The diseases are not restricted to Finns; they are genetic diseases with far wider distribution in the world, but due to founder effects and genetic isolation they are more common in Finns.

Lujan–Fryns syndrome (LFS) is an X-linked genetic disorder that causes mild to moderate intellectual disability and features described as Marfanoid habitus, referring to a group of physical characteristics similar to those found in Marfan syndrome. These features include a tall, thin stature and long, slender limbs. LFS is also associated with psychopathology and behavioral abnormalities, and it exhibits a number of malformations affecting the brain and heart. The disorder is inherited in an X-linked dominant manner, and is attributed to a missense mutation in the MED12 gene. There is currently no treatment or therapy for the underlying MED12 malfunction, and the exact cause of the disorder remains unclear.
Malpuech facial clefting syndrome, also called Malpuech syndrome or Gypsy type facial clefting syndrome, is a rare congenital syndrome. It is characterized by facial clefting, a caudal appendage, growth deficiency, intellectual and developmental disability, and abnormalities of the renal system (kidneys) and the male genitalia. Abnormalities of the heart, and other skeletal malformations may also be present. The syndrome was initially described by Georges Malpuech and associates in 1983. It is thought to be genetically related to Juberg-Hayward syndrome. Malpuech syndrome has also been considered as part of a spectrum of congenital genetic disorders associated with similar facial, urogenital and skeletal anomalies. Termed "3MC syndrome", this proposed spectrum includes Malpuech, Michels and Mingarelli-Carnevale (OSA) syndromes. Mutations in the COLLEC11 and MASP1 genes are believed to be a cause of these syndromes. The incidence of Malpuech syndrome is unknown. The pattern of inheritance is autosomal recessive, which means a defective (mutated) gene associated with the syndrome is located on an autosome, and the syndrome occurs when two copies of this defective gene are inherited.
Nasodigitoacoustic syndrome, also called Keipert syndrome, is a rare congenital syndrome first described by J.A. Keipert and colleagues in 1973. The syndrome is characterized by a misshaped nose, broad thumbs and halluces, brachydactyly, sensorineural hearing loss, facial features such as hypertelorism, and developmental delay. It is believed to be inherited in an X-linked recessive manner, which means a genetic mutation causing the disorder is located on the X chromosome, and while two copies of the mutated gene must be inherited for a female to be born with the disorder, just one copy is sufficient to cause a male to be born with the disorder. Nasodigitoacoustic syndrome is likely caused by a mutated gene located on the X chromosome between positions Xq22.2–q28. The incidence of the syndrome has not been determined, but it is considered to affect less than 200,000 people in the United States, and no greater than 1 per 2,000 in Europe. It is similar to Keutel, Muenke, Rubinstein and Teunissen-Cremers syndrome.
Floating–Harbor syndrome, also known as Pelletier–Leisti syndrome, is a rare disease with fewer than 50 cases described in the literature. It is usually diagnosed in early childhood and is characterized by the triad of proportionate short stature with delayed bone age, characteristic facial appearance, and delayed speech development. Although its cause is unknown, it is thought to result from genetic mutation, and diagnosis is established by the presence of a heterozygous SRCAP mutation in those with clinical findings of FHS.
Liebenberg Syndrome is a rare autosomal genetic disease that involves a deletion mutation upstream of the PITX1 gene, which is one that's responsible for the body's organization, specifically in forming lower limbs. In animal studies, when this deletion was introduced to developing birds, their wing buds were noted to take on limb-like structures.
Primary juvenile glaucoma is glaucoma that develops due to ocular hypertension and is evident either at birth or within the first few years of life. It is caused due to abnormalities in the anterior chamber angle development that obstruct aqueous outflow in the absence of systemic anomalies or other ocular malformation.

BARX homeobox 1 is a protein that in humans is encoded by the BARX1 gene.