An intermolecular force (IMF) is the force that mediates interaction between molecules, including the electromagnetic forces of attraction or repulsion which act between atoms and other types of neighbouring particles, e.g. atoms or ions. Intermolecular forces are weak relative to intramolecular forces – the forces which hold a molecule together. For example, the covalent bond, involving sharing electron pairs between atoms, is much stronger than the forces present between neighboring molecules. Both sets of forces are essential parts of force fields frequently used in molecular mechanics.
The van der Waals radius, rw, of an atom is the radius of an imaginary hard sphere representing the distance of closest approach for another atom. It is named after Johannes Diderik van der Waals, winner of the 1910 Nobel Prize in Physics, as he was the first to recognise that atoms were not simply points and to demonstrate the physical consequences of their size through the van der Waals equation of state.

In molecular physics, the van der Waals force is a distance-dependent interaction between atoms or molecules. Unlike ionic or covalent bonds, these attractions do not result from a chemical electronic bond; they are comparatively weak and therefore more susceptible to disturbance. The van der Waals force quickly vanishes at longer distances between interacting molecules.

In chemistry and thermodynamics, the Van der Waals equation is an equation of state which extends the ideal gas law to include the effects of interaction between molecules of a gas, as well as accounting for the finite size of the molecules.
Density-functional theory (DFT) is a computational quantum mechanical modelling method used in physics, chemistry and materials science to investigate the electronic structure of many-body systems, in particular atoms, molecules, and the condensed phases. Using this theory, the properties of a many-electron system can be determined by using functionals, i.e. functions of another function. In the case of DFT, these are functionals of the spatially dependent electron density. DFT is among the most popular and versatile methods available in condensed-matter physics, computational physics, and computational chemistry.

Assisted Model Building with Energy Refinement (AMBER) is a family of force fields for molecular dynamics of biomolecules originally developed by Peter Kollman's group at the University of California, San Francisco. AMBER is also the name for the molecular dynamics software package that simulates these force fields. It is maintained by an active collaboration between David Case at Rutgers University, Tom Cheatham at the University of Utah, Adrian Roitberg at University of Florida, Ken Merz at Michigan State University, Carlos Simmerling at Stony Brook University, Ray Luo at UC Irvine, and Junmei Wang at Encysive Pharmaceuticals.

London dispersion forces are a type of intermolecular force acting between atoms and molecules that are normally electrically symmetric; that is, the electrons are symmetrically distributed with respect to the nucleus. They are part of the van der Waals forces. The LDF is named after the German physicist Fritz London. They are the weakest intermolecular force.

The Stark effect is the shifting and splitting of spectral lines of atoms and molecules due to the presence of an external electric field. It is the electric-field analogue of the Zeeman effect, where a spectral line is split into several components due to the presence of the magnetic field. Although initially coined for the static case, it is also used in the wider context to describe the effect of time-dependent electric fields. In particular, the Stark effect is responsible for the pressure broadening of spectral lines by charged particles in plasmas. For most spectral lines, the Stark effect is either linear or quadratic with a high accuracy.
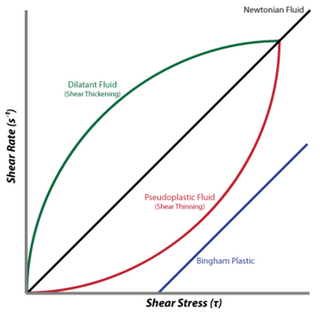
A dilatant material is one in which viscosity increases with the rate of shear strain. Such a shear thickening fluid, also known by the initialism STF, is an example of a non-Newtonian fluid. This behaviour is usually not observed in pure materials, but can occur in suspensions.
The DLVO theory explains the aggregation and kinetic stability of aqueous dispersions quantitatively and describes the force between charged surfaces interacting through a liquid medium. It combines the effects of the van der Waals attraction and the electrostatic repulsion due to the so-called double layer of counterions. The electrostatic part of the DLVO interaction is computed in the mean field approximation in the limit of low surface potentials - that is when the potential energy of an elementary charge on the surface is much smaller than the thermal energy scale, . For two spheres of radius each having a charge separated by a center-to-center distance in a fluid of dielectric constant containing a concentration of monovalent ions, the electrostatic potential takes the form of a screened-Coulomb or Yukawa potential,
In differential geometry, the Laplace–Beltrami operator is a generalization of the Laplace operator to functions defined on submanifolds in Euclidean space and, even more generally, on Riemannian and pseudo-Riemannian manifolds. It is named after Pierre-Simon Laplace and Eugenio Beltrami.
The Hückel method or Hückel molecular orbital theory, proposed by Erich Hückel in 1930, is a simple method for calculating molecular orbitals as linear combinations of atomic orbitals. The theory predicts the molecular orbitals for π-electrons in π-delocalized molecules, such as ethylene, benzene, butadiene, and pyridine. It provides the theoretical basis for Hückel's rule that cyclic, planar molecules or ions with π-electrons are aromatic. It was later extended to conjugated molecules such as pyridine, pyrrole and furan that contain atoms other than carbon and hydrogen (heteroatoms). A more dramatic extension of the method to include σ-electrons, known as the extended Hückel method (EHM), was developed by Roald Hoffmann. The extended Hückel method gives some degree of quantitative accuracy for organic molecules in general and was used to provide computational justification for the Woodward–Hoffmann rules. To distinguish the original approach from Hoffmann's extension, the Hückel method is also known as the simple Hückel method (SHM).
In mathematics, the Loomis–Whitney inequality is a result in geometry, which in its simplest form, allows one to estimate the "size" of a -dimensional set by the sizes of its -dimensional projections. The inequality has applications in incidence geometry, the study of so-called "lattice animals", and other areas.
In molecular physics, the Hamaker constant is a physical constant that can be defined for a van der Waals (vdW) body–body interaction:
In statistical mechanics, the cluster expansion is a power series expansion of the partition function of a statistical field theory around a model that is a union of non-interacting 0-dimensional field theories. Cluster expansions originated in the work of Mayer & Montroll (1941). Unlike the usual perturbation expansion which usually leads to a divergent asymptotic series, the cluster expansion may converge within a non-trivial region, in particular when the interaction is small and short-ranged.
Chapman–Enskog theory provides a framework in which equations of hydrodynamics for a gas can be derived from the Boltzmann equation. The technique justifies the otherwise phenomenological constitutive relations appearing in hydrodynamical descriptions such as the Navier–Stokes equations. In doing so, expressions for various transport coefficients such as thermal conductivity and viscosity are obtained in terms of molecular parameters. Thus, Chapman–Enskog theory constitutes an important step in the passage from a microscopic, particle-based description to a continuum hydrodynamical one.

The feet of geckos have a number of specializations. Their surfaces can adhere to any type of material with the exception of Teflon (PTFE). This phenomenon can be explained with three elements:
In computational chemistry and molecular dynamics, the combination rules or combining rules are equations that provide the interaction energy between two dissimilar non-bonded atoms, usually for the part of the potential representing the van der Waals interaction. In the simulation of mixtures, the choice of combining rules can sometimes affect the outcome of the simulation.
In condensed matter physics and physical chemistry, the Lifshitz theory of van der Waals forces, sometimes called the macroscopic theory of van der Waals forces, is a method proposed by Evgeny Mikhailovich Lifshitz in 1954 for treating van der Waals forces between bodies which does not assume pairwise additivity of the individual intermolecular forces; that is to say, the theory takes into account the influence of neighboring molecules on the interaction between every pair of molecules located in the two bodies, rather than treating each pair independently.


