Related Research Articles

Proteolysis is the breakdown of proteins into smaller polypeptides or amino acids. Uncatalysed, the hydrolysis of peptide bonds is extremely slow, taking hundreds of years. Proteolysis is typically catalysed by cellular enzymes called proteases, but may also occur by intra-molecular digestion.

A protease is an enzyme that catalyzes proteolysis, breaking down proteins into smaller polypeptides or single amino acids, and spurring the formation of new protein products. They do this by cleaving the peptide bonds within proteins by hydrolysis, a reaction where water breaks bonds. Proteases are involved in many biological functions, including digestion of ingested proteins, protein catabolism, and cell signaling.

Post-translational modification (PTM) is the covalent process of changing proteins following protein biosynthesis. PTMs may involve enzymes or occur spontaneously. Proteins are created by ribosomes translating mRNA into polypeptide chains, which may then change to form the mature protein product. PTMs are important components in cell signalling, as for example when prohormones are converted to hormones.
A metalloproteinase, or metalloprotease, is any protease enzyme whose catalytic mechanism involves a metal. An example is ADAM12 which plays a significant role in the fusion of muscle cells during embryo development, in a process known as myogenesis.

A calpain is a protein belonging to the family of calcium-dependent, non-lysosomal cysteine proteases expressed ubiquitously in mammals and many other organisms. Calpains constitute the C2 family of protease clan CA in the MEROPS database. The calpain proteolytic system includes the calpain proteases, the small regulatory subunit CAPNS1, also known as CAPN4, and the endogenous calpain-specific inhibitor, calpastatin.
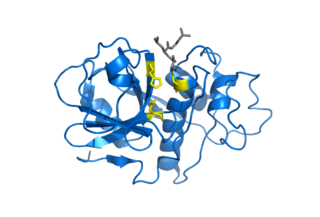
Cysteine proteases, also known as thiol proteases, are hydrolase enzymes that degrade proteins. These proteases share a common catalytic mechanism that involves a nucleophilic cysteine thiol in a catalytic triad or dyad.
In molecular biology, the Signal Peptide Peptidase (SPP) is a type of protein that specifically cleaves parts of other proteins. It is an intramembrane aspartyl protease with the conserved active site motifs 'YD' and 'GxGD' in adjacent transmembrane domains (TMDs). Its sequences is highly conserved in different vertebrate species. SPP cleaves remnant signal peptides left behind in membrane by the action of signal peptidase and also plays key roles in immune surveillance and the maturation of certain viral proteins.
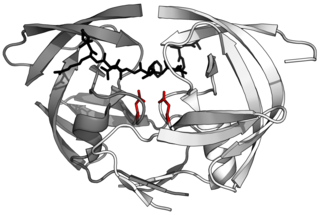
Aspartic proteases are a catalytic type of protease enzymes that use an activated water molecule bound to one or more aspartate residues for catalysis of their peptide substrates. In general, they have two highly conserved aspartates in the active site and are optimally active at acidic pH. Nearly all known aspartyl proteases are inhibited by pepstatin.
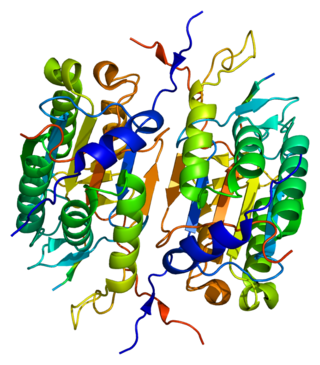
Caspase 2 also known as CASP2 is an enzyme that, in humans, is encoded by the CASP2 gene. CASP2 orthologs have been identified in nearly all mammals for which complete genome data are available. Unique orthologs are also present in birds, lizards, lissamphibians, and teleosts.
Caspase 4 is an enzyme that proteolytically cleaves other proteins at an aspartic acid residue (LEVD-), and belongs to a family of cysteine proteases called caspases. The function of caspase 4 is not fully known, but it is believed to be an inflammatory caspase, along with caspase 1, caspase 5, with a role in the immune system.
MEROPS is an online database for peptidases and their inhibitors. The classification scheme for peptidases was published by Rawlings & Barrett in 1993, and that for protein inhibitors by Rawlings et al. in 2004. The most recent version, MEROPS 12.4, was released in late October 2021.

Caspase-7, apoptosis-related cysteine peptidase, also known as CASP7, is a human protein encoded by the CASP7 gene. CASP7 orthologs have been identified in nearly all mammals for which complete genome data are available. Unique orthologs are also present in birds, lizards, lissamphibians, and teleosts.

ATP-dependent Clp protease proteolytic subunit (ClpP) is an enzyme that in humans is encoded by the CLPP gene. This protein is an essential component to form the protein complex of Clp protease.
The Proteolysis MAP (PMAP) was an integrated web resource focused on proteases. Its domain now links to a scam/spam browser extender.

In molecular biology short linear motifs (SLiMs), linear motifs or minimotifs are short stretches of protein sequence that mediate protein–protein interaction.

Threonine proteases are a family of proteolytic enzymes harbouring a threonine (Thr) residue within the active site. The prototype members of this class of enzymes are the catalytic subunits of the proteasome, however the acyltransferases convergently evolved the same active site geometry and mechanism.
Terminal amine isotopic labeling of substrates (TAILS) is a method in quantitative proteomics that identifies the protein content of samples based on N-terminal fragments of each protein and detects differences in protein abundance among samples.

Fast parallel proteolysis (FASTpp) is a method to determine the thermostability of proteins by measuring which fraction of protein resists rapid proteolytic digestion.
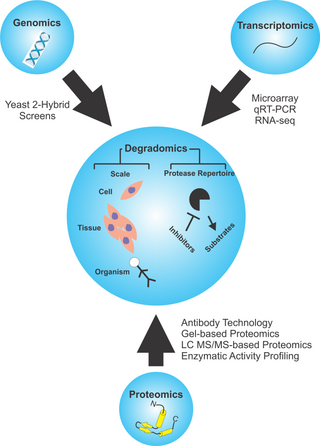
Degradomics is a sub-discipline of biology encompassing all the genomic and proteomic approaches devoted to the study of proteases, their inhibitors, and their substrates on a system-wide scale. This includes the analysis of the protease and protease-substrate repertoires, also called "protease degradomes". The scope of these degradomes can range from cell, tissue, and organism-wide scales.
Asparagine peptide lyase are one of the seven groups in which proteases, also termed proteolytic enzymes, peptidases, or proteinases, are classified according to their catalytic residue. The catalytic mechanism of the asparagine peptide lyases involves an asparagine residue acting as nucleophile to perform a nucleophilic elimination reaction, rather than hydrolysis, to catalyse the breaking of a peptide bond.
References
- 1 2 Lange, P. F.; Huesgen, P. F.; Overall, C. M. (2011). "TopFIND 2.0--linking protein termini with proteolytic processing and modifications altering protein function". Nucleic Acids Research. 40 (Database issue): D351–D361. doi:10.1093/nar/gkr1025. PMC 3244998 . PMID 22102574.
- ↑ Lange, P. F.; Overall, C. M. (2011). "TopFIND, a knowledgebase linking protein termini with function". Nature Methods. 8 (9): 703–704. doi:10.1038/nmeth.1669. PMID 21822272. S2CID 7195106.
- ↑ Cox JH, Starr AE, Kappelhoff R, Yan R, Roberts CR, Overall CM (December 2010). "Matrix metalloproteinase 8 deficiency in mice exacerbates inflammatory arthritis through delayed neutrophil apoptosis and reduced caspase 11 expression". Arthritis & Rheumatism. 62 (12): 3645–3655. doi:10.1002/art.27757. PMID 21120997.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ↑ Overall CM, Kleifeld O (March 2006). "Tumour microenvironment - opinion: validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy". Nature Reviews Cancer. 6 (3): 227–239. doi:10.1038/nrc1821. PMID 16498445. S2CID 21114447.
- ↑ Nikolaus Fortelny, Jennifer H. Cox, Reinhild Kappelhoff, Amanda E. Starr, Philipp F. Lange, Paul Pavlidis & Christopher M. Overall (2014). "Network analyses reveal pervasive functional regulation between proteases in the human protease web". PLOS Biology . 12 (5): e1001869. doi: 10.1371/journal.pbio.1001869 . PMC 4035269 . PMID 24865846.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ↑ Nikolaus Fortelny, Georgina S. Butler, Christopher M. Overall & Paul Pavlidis (2017). "Protease-Inhibitor Interaction Predictions: Lessons on the Complexity of Protein-Protein Interactions". Molecular & Cellular Proteomics . 16 (6): 1038–1051. doi:10.1074/mcp.M116.065706. PMC 5461536 . PMID 28385878.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ↑ Pitter F. Huesgen, Philipp F. Lange & Christopher M. Overall (2014). "Ensembles of protein termini and specific proteolytic signatures as candidate biomarkers of disease". Proteomics: Clinical Applications . 8 (5–6): 338–350. doi:10.1002/prca.201300104. PMID 24497460. S2CID 24591183.
- ↑ Philipp F. Lange & Christopher M. Overall (2013). "Protein TAILS: when termini tell tales of proteolysis and function". Current Opinion in Chemical Biology . 17 (1): 73–82. doi: 10.1016/j.cbpa.2012.11.025 . PMID 23298954.
- ↑ Philipp F. Lange, Pitter F. Huesgen, Karen Nguyen & Christopher M. Overall (2014). "Annotating N termini for the human proteome project: N termini and Nalpha-acetylation status differentiate stable cleaved protein species from degradation remnants in the human erythrocyte proteome". Journal of Proteome Research . 13 (4): 2028–2044. doi:10.1021/pr401191w. PMC 3979129 . PMID 24555563.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ↑ Nikolaus Fortelny, Sharon Yang, Paul Pavlidis, Philipp F. Lange & Christopher M. Overall (2015). "Proteome TopFIND 3.0 with TopFINDer and PathFINDer: database and analysis tools for the association of protein termini to pre- and post-translational events". Nucleic Acids Research . 43 (Database issue): D290–D297. doi:10.1093/nar/gku1012. PMC 4383881 . PMID 25332401.
{{cite journal}}: CS1 maint: multiple names: authors list (link)