Trypanosoma brucei is a species of parasitic kinetoplastid belonging to the genus Trypanosoma that is present in sub-Saharan Africa. Unlike other protozoan parasites that normally infect blood and tissue cells, it is exclusively extracellular and inhabits the blood plasma and body fluids.[1] It causes deadly vector-borne diseases: African trypanosomiasis or sleeping sickness in humans, and animal trypanosomiasis or nagana in cattle and horses.[2] It is a species complex grouped into three subspecies: T. b. brucei, T. b. gambiense and T. b. rhodesiense.[3] The first is a parasite of non-human mammals and causes nagana, while the latter two are zoonotic infecting both humans and animals and cause African trypanosomiasis.
T. brucei is transmitted between mammal hosts by an insectvector belonging to different species of tsetse fly (Glossina). Transmission occurs by biting during the insect's blood meal. The parasites undergo complex morphological changes as they move between insect and mammal over the course of their life cycle. The mammalian bloodstream forms are notable for their cell surface proteins, variant surface glycoproteins, which undergo remarkable antigenic variation, enabling persistent evasion of host adaptive immunity leading to chronic infection. T. brucei is one of only a few pathogens known to cross the blood-brain barrier.[4] There is an urgent need for the development of new drug therapies, as current treatments can have severe side effects and can prove fatal to the patient.[5]
Whilst not historically regarded as T. brucei subspecies due to their different means of transmission, clinical presentation, and loss of kinetoplast DNA, genetic analyses reveal that T. equiperdum and T. evansi are evolved from parasites very similar to T. b. brucei, and are thought to be members of the bruceiclade.[6]
The parasite was discovered in 1894 by Sir David Bruce, after whom the scientific name was given in 1899.[7][8]
History and discovery
Early records
Sleeping sickness in animals was described in ancient Egyptian writings. During the Middle Ages, Arabian traders noted the prevalence of sleeping sickness among Africans and their dogs.[9] It was a major infectious disease in southern and eastern Africa in the 19th century.[10] The Zulu Kingdom (now part of South Africa) was severely struck by the disease, which became known to the British as nagana,[2] a Zulu word meaning "low or depressed in spirit." In other parts of Africa, Europeans called it the "fly disease."[11][12]
A man having sleeping sickness at Buruma Island, Uganda.
John Aktins, an English naval surgeon, gave the first medical description of human sleeping sickness in 1734. He attributed deaths, which he called "sleepy distemper," in Guinea to the infection.[13] Another English physician, Thomas Masterman Winterbottom, gave a clearer description of the symptoms from Sierra Leone in 1803.[14] Winterbottom described a key feature of the disease, swollen posterior cervical lymph nodes, and slaves who developed such swellings were ruled unfit for trade.[13] The symptom is eponymously known as "Winterbottom's sign".[15]
Discovery of the parasite
The Royal Army Medical Corps appointed David Bruce, who at the time was assistant professor of pathology at the Army Medical School in Netley with a rank of Captain in the army, in 1894 to investigate a disease known as nagana in South Africa. The disease caused severe problems among the local cattle and British Army horses.[3] On 27 October 1894, Bruce and his microbiologist-wife Mary Elizabeth Bruce (née Steele) moved to Ubombo Hill, where the disease was most prevalent.[16]
On the sixth day of investigation, Bruce identified parasites from the blood of diseased cows. He initially noted them as a kind of filaria (tiny roundworms), but by the end of the year established that the parasites were "haematozoa" (protozoan) and were the cause of nagana.[3] It was the discovery of Trypanosoma brucei.[17] The scientific name was created by British zoologists Henry George Plimmer and John Rose Bradford in 1899 as Trypanosoma brucii due to printer's error.[3][18] The genus Trypanosoma was already introduced by Hungarian physician David Gruby in his description of T. sanguinis, a species he discovered in frogs in 1843.[19]
Outbreaks
In Uganda, the first case of human infection was reported in 1898.[10] It was followed by an outbreak in 1900.[20] By 1901, it became severe with death toll estimated to about 20,000.[21] More than 250,000 people died in the epidemic that lasted for two decades.[20] The disease commonly popularised as "negro lethargy."[22][23] It was not known whether the human sleeping sickness and nagana were similar or the two disease were caused by similar parasites.[24] Even the observations of Forde and Dutton did not indicate that the trypanosome was related to sleeping sickness.[25]
The Royal Society constituted a three-member Sleeping Sickness Commission on 10 May 1902 to investigate the epidemic in Uganda.[26] The Commission comprised George Carmichael Low from the London School of Hygiene and Tropical Medicine as the leader, his colleague Aldo Castellani and Cuthbert Christy, a medical officer on duty in Bombay, India.[27][28] At the time, a debate remained on the etiology, some favoured bacterial infection while some believed as helminth infection.[29] The first investigation focussed on Filaria perstans (later renamed Mansonella perstans), a small roundworm transmitted by flies, and bacteria as possible causes, only to discover that the epidemic was not related to these pathogens.[30][31] The team was described as an "ill-assorted group"[31] and a "queer lot",[32] and the expedition "a failure."[21] Low, whose conduct was described as "truculent and prone to take offence," left the Commission and Africa after three months.[33]
In February 1902, the British War Office, following a request from the Royal Society, appointed David Bruce to lead the second Sleeping Sickness Commission.[34] With David Nunes Nabarro (from the University College Hospital), Bruce and his wife joined Castellani and Christy on 16 March.[31] In November 1902, Castellani had found the trypanosomes in the cerebrospinal fluid of an infected person. He was convinced that the trypanosome was the causative parasite of sleeping sickness. Like Low, his conduct has been criticised and the Royal Society refused to publish his report. He was further infuriated when Bruce advised him not to make rash conclusion without further evidences, as there were many other parasites to consider.[26] Castellani left Africa in April and published his report as "On the discovery of a species of Trypanosoma in the cerebrospinal fluid of cases of sleeping sickness" in The Lancet.[35] By then the Royal Society had already published the report.[36] By August 1903, Bruce and his team established that the disease was transmitted by the tsetse fly, Glossina palpalis.[37] However, Bruce did not understand the trypanosoma life cycle and believed that the parasites were simply transmitted from one person to another.[9]
Around the same time, Germany sent an expeditionary team led by Robert Koch to investigate the epidemic in Togo and East Africa. In 1909, one of the team members, Friedrich Karl Kleine discovered that the parasite had developmental stages in the tsetse flies.[9] Bruce, in the third Sleeping Sickness Commission (1908–1912) that included Albert Ernest Hamerton, H.R. Bateman and Frederick Percival Mackie, established the basic developmental cycle through which the trypanosome in tsetse fly must pass.[38][39] An open question, noted by Bruce at this stage, was how the trypanosome finds its way to the salivary glands. Muriel Robertson,[40][41] in experiments carried out between 1911 and 1912, established how ingested trypanosomes finally reach the salivary glands of the fly.
Discovery of human trypanosomes
British Colonial Surgeon Robert Michael Forde was the first to find the parasite in human. He found it from an English steamboat captain who was admitted to a hospital at Bathurst, Gambia, in 1901.[9] His report in 1902 indicates that he believed it to be a kind of filarial worm.[14] From the same person, Forde's colleague Joseph Everett Dutton identified it as a protozoan belonging to the genus Trypanosoma.[3] Knowing the distinct features, Dutton proposed a new species name in 1902:
At present then it is impossible to decide definitely as to the species, but if on further study it should be found to differ from other disease-producing trypanosomes I would suggest that it be called Trypanosoma gambiense.[42]
Another human trypanosome (now called T. brucei rhodesiense) was discovered by British parasitologists John William Watson Stephens and Harold Benjamin Fantham.[9] In 1910, Stephens noted in his experimental infection in rats that the trypanosome, obtained from an individual from Northern Rhodesia (later Zambia), was not the same as T. gambiense. The source of the parasite, an Englishman travelling in Rhodesia was found with the blood parasites in 1909, and was transported to and admitted at the Royal Southern Hospital in Liverpool under the care of Ronald Ross.[3] Fantham described the parasite's morphology and found that it was a different trypanosome.[43][44]
Species
T. brucei is a species complex that includes:
T. brucei gambiense which causes slow onset chronic trypanosomiasis in humans. It is most common in central and western Africa, where humans are thought to be the primary reservoir.[45] In 1973, David Hurst Molyneux was the first to find infection of this strain in wildlife and domestic animals.[46][47] Since 2002, there are several reports showing that animals, including cattle, are also infected.[47] It is responsible for 98% of all human African trypanosomiasis,[48] and is roughly 100% fatal when left untreated.[49]
T. brucei rhodesiense which causes fast onset acute trypanosomiasis in humans. A highly zoonotic parasite, it is prevalent in southern and eastern Africa, where game animals and livestock are thought to be the primary reservoir.[45][48]
T. brucei brucei which causes animal trypanosomiasis, along with several other species of Trypanosoma. T. b. brucei is not infective to humans due to its susceptibility to lysis by trypanosome lytic factor-1 (TLF-1).[50][51] However, it is closely related to, and shares fundamental features with the human-infective subspecies.[52] Only rarely can the T. b. brucei infect a human.[53]
The subspecies cannot be distinguished from their structure as they are all identical under microscopes. Geographical location is the main distinction.[48] Molecular markers have been developed for individual identification. Serum resistance-associated (SRA) gene is used to differentiate T. b. rhodesiense from other subspecies.[54]TgsGP gene, found only in type 1 T. b. gambiense is also a specific distinction between T. b. gambiense strains.[55]
The subspecies lack many of the features commonly considered necessary to constitute monophyly.[56] As such Lukeš et al., 2022 proposes a new polyphyly by ecotype.[56]
Etymology
The genus name is derived from two Greek words: τρυπανον (trypanon or trupanon), which means "borer" or "auger", referring to the corkscrew-like movement;[57] and σῶμα (sôma), meaning "body."[58][59] The specific name is after David Bruce, who discovered the parasites in 1894.[7][8] The subspecies, the human strains, are named after the regions in Africa where they were first identified: T. brucei gambiense was described from an Englishman in Gambia in 1901; T. brucei rhodesiense was found from another Englishman in Northern Rhodesia in 1909.[3]
Structure
SEM false colour micrograph of the procyclic form as found in the tsetse fly midgut. The cell body is shown in orange and the flagellum is in red. 84 pixels/μm.
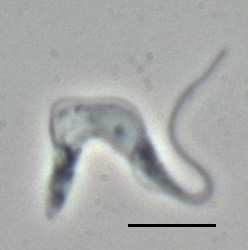
T. brucei is a typical unicellular eukaryotic cell, and measures 8 to 50 μm in length. It has an elongated body having a streamlined and tapered shape. Its cell membrane (called pellicle) encloses the cell organelles, including the nucleus, mitochondria, endoplasmic reticulum, Golgi apparatus, and ribosomes. In addition, there is an unusual organelle called the kinetoplast, which is a complex of thousands of interlinked circles of mitochondrial DNA known as mini- and maxicircles.[60] The kinetoplast lies near the basal body with which it is indistinguishable under microscope. From the basal body arises a single flagellum that run towards the anterior end. Along the body surface, the flagellum is attached to the cell membrane forming an undulating membrane. Only the tip of the flagellum is free at the anterior end.[61] The cell surface of the bloodstream form features a dense coat of variant surface glycoproteins (VSGs) which is replaced by an equally dense coat of procyclins when the parasite differentiates into the procyclic phase in the tsetse fly midgut.[62]
The six main morphologies of trypanosomatids. The different life cycle stages of T brucei fall into the trypomastigote and epimastigote morphological categories.
Trypanosomatids show several different classes of cellular organisation of which two are adopted by T. brucei at different stages of the life cycle:[61]
Epimastigote, which is found in tsetse fly. Its kinetoplast and basal body lie anterior to the nucleus, with a long flagellum attached along the cell body. The flagellum starts from the centre of the body.
Trypomastigote, which is found in mammalian hosts. The kinetoplast and basal body are posterior of nucleus. The flagellum arises from the posterior end of the body.
Flagellar structure
These names are derived from the Greekmastig- meaning whip, referring to the trypanosome's whip-like flagellum. The trypanosome flagellum has two main structures. It is made up of a typical flagellar axoneme, which lies parallel to the paraflagellar rod,[63] a lattice structure of proteins unique to the kinetoplastids, euglenoids and dinoflagellates.[64][65]
The microtubules of the flagellar axoneme lie in the normal 9+2 arrangement, orientated with the + at the anterior end and the − in the basal body. The cytoskeletal structure extends from the basal body to the kinetoplast. The flagellum is bound to the cytoskeleton of the main cell body by four specialised microtubules, which run parallel and in the same direction to the flagellar tubulin.[66][67]
The flagellar function is twofold — locomotion via oscillations along the attached flagellum and cell body in human blood stream and tsetse fly gut,[68][69] and attachment to the salivary gland epithelium of the fly during the epimastigote stage.[57][70] The flagellum propels the body in such a way that the axoneme generates the oscillation and a flagellar wave is created along the undulating membrane. As a result, the body moves in a corkscrew pattern.[57] In flagella of other organisms, the movement starts from the base towards the tip, while in T. brucei and other trypanosomatids, the beat originates from the tip and progresses towards the base, forcing the body to move towards the direction of the tip of the flagellum.[70]
Life cycle
Life cycle
T. brucei completes its life cycle between tsetse fly (of the genus Glossina) and mammalian hosts, including humans, cattle, horses, and wild animals. In stressful environments, T. brucei produces exosomes containing the spliced leader RNA and uses the endosomal sorting complexes required for transport (ESCRT) system to secrete them as extracellular vesicles.[71] When absorbed by other trypanosomes these EVs cause repulsive movement away from the area and so away from bad environments.[71]
In mammalian host
Infection occurs when a vector tsetse fly bites a mammalian host. The fly injects the metacyclic trypomastigotes into the skin tissue. The trypomastigotes enter the lymphatic system and into the bloodstream. The initial trypomastigotes are short and stumpy (SS).[72] They are protected from the host's immune system by producing antigentic variation called variant surface glycoproteins on their body surface.[1] Once inside the bloodstream, they grow into long and slender forms (LS). Then, they multiply by binary fission. Some of the daughter cells then become short and stumpy again.[73][74][2] Some of them remains as intermediate forms, representing a transitional stage between the long and short forms.[48] The long slender forms are able to penetrate the blood vessel endothelium and invade extravascular tissues, including the central nervous system (CNS)[68] and placenta in pregnant women.[75]
Sometimes, wild animals can be infected by the tsetse fly and they act as reservoirs. In these animals, they do not produce the disease, but the live parasite can be transmitted back to the normal hosts.[72] Besides preparation to be taken up and vectored to another host by a tsetse fly, transition from LS to SS in the mammal serves to prolong the host's lifespan – controlling parasitemia aids in increasing the total transmitting duration of any particular infested host.[74][2]
In tsetse fly
Unlike anopheline mosquitos and sandflies that transmit other protozoan infections in which only females are involved, both sexes of tsetse flies are blood feeders and equally transmit trypanosomes.[76] The short and stumpy trypomastigotes (SS) are taken up by tsetse flies during a blood meal.[74][2] Survival in the tsetse midgut is one reason for the particular adaptations of the SS stage.[74][2] The trypomastigotes enter the midgut of the fly where they become procyclic trypomastigotes as they replace their VSG with other protein coats called procyclins.[1] Because the fly faces digestive damage from immune factors in the bloodmeal, it produces serpins to suppress the infection. The serpins including GmmSRPN3, GmmSRPN5, GmmSRPN9, and especially GmmSRPN10 are then hijacked by the parasite to aid its own midgut infection, using them to inactivate bloodmeal trypanolytic factors which would otherwise make the fly host inhospitable.[77]:346
The procyclic trypomastigotes cross the peritrophic matrix, undergo slight elongation and migrate to the anterior part of the midgut as non-proliferative long mesocyclic trypomastigotes. As they reach the proventriculus, they became thinner and undergo cytoplasmic rearrangement to give rise to proliferative epimastigotes.[76] The epimastigotes divide asymmetrically to produce long and short epimastigotes. The long epimastigote cannot move to other places and simply die off by apoptosis.[78][79] The short epimastigote migrate from the proventriculus via the foregut and proboscis to the salivary glands where they get attached to the salivary gland epithelium.[57] Even all the short forms do not succeed in the complete migration to the salivary glands as most of them perish on the way–only up to five may survive.[76][80]
In the salivary glands, the survivors undergo phases of reproduction. The first cycle in an equal mitosis by which a mother cell produces two similar daughter epimastigotes. They remain attach to the epithelium. This phase is the main reproduction in first-stage infection to ensure sufficient number of parasites in the salivary gland.[76] The second cycle, which usually occurs in late-stage infection, involves unequal mitosis that produces two different daughter cells from the mother epimastigote. One daughter is an epimastigote that remains non-infective and the other is a trypomastigote.[81] The trypomastigote detach from the epithelium and undergo transformation into short and stumpy trypomastigotes. The surface procyclins are replaced with VSGs and become the infective metacyclic trypomastigotes.[48] Complete development in the fly takes about 20 days.[72][73] They are injected into the mammalian host along with the saliva on biting, and are known as salivarian.[76]
In the case of T. b. brucei infecting Glossina palpalis gambiensis, the parasite changes the proteome contents of the fly's head and causes behavioral changes such as unnecessarily increased feeding frequency, which increases transmission opportunities. This is related to altered glucose metabolism that causes a perceived need for more calories. (The metabolic change, in turn, being due to complete absence of glucose-6-phosphate 1-dehydrogenase in infected flies.) Monoamine neurotransmitter synthesis is also altered: production of aromatic L-amino acid decarboxylase involved in dopamine and serotonin synthesis, and α-methyldopa hypersensitive protein was induced. This is similar to the alterations in other dipteran vectors' head proteomes under infection by other eukaryotic parasites of mammals.[82]
Reproduction
Binary fission
Trypanosome cell cycle (procyclic form)
The reproduction of T. brucei is unusual compared to most eukaryotes. The nuclear membrane remains intact and the chromosomes do not condense during mitosis. The basal body, unlike the centrosome of most eukaryotic cells, does not play a role in the organisation of the spindle and instead is involved in division of the kinetoplast. The events of reproduction are:[61]
The basal body duplicates and both remain associated with the kinetoplast. Each basal body forms a separate flagellum.
Kinetoplast DNA undergoes synthesis then the kinetoplast divides coupled with separation of the two basal bodies.
Nuclear DNA undergoes synthesis while a new flagellum extends from the younger, more posterior, basal body.
The nucleus undergoes mitosis.
Cytokinesis progresses from the anterior to posterior.
In the 1980s, DNA analyses of the developmental stages of T. brucei started to indicate that the trypomastigote in the tsetse fly undergoes meiosis, i.e., a sexual reproduction stage.[83] But it is not always necessary for a complete life cycle.[84] The existence of meiosis-specific proteins was reported in 2011.[85] The haploid gametes (daughter cells produced after meiosis) were discovered in 2014. The haploid trypomastigote-like gametes can interact with each other via their flagella and undergo cell fusion (the process is called syngamy).[86][87] Thus, in addition to binary fission, T. brucei can multiply by sexual reproduction. Trypanosomes belong to the supergroup Excavata and are one of the earliest diverging lineages among eukaryotes.[88] The discovery of sexual reproduction in T. brucei supports the hypothesis that meiosis and sexual reproduction are ancestral and ubiquitous features of eukaryotes.[89]
The insect vectors for T. brucei are different species of tsetse fly (genus Glossina). The major vectors of T. b. gambiense, causing West African sleeping sickness, are G. palpalis, G. tachinoides, and G. fuscipes. While the principal vectors of T. b. rhodesiense, causing East African sleeping sickness, are G. morsitans, G. pallidipes, and G. swynnertoni. Animal trypanosomiasis is transmitted by a dozen species of Glossina.[90]
In later stages of a T. brucei infection of a mammalian host the parasite may migrate from the bloodstream to also infect the lymph and cerebrospinal fluids. It is under this tissue invasion that the parasites produce the sleeping sickness.[72]
In addition to the major form of transmission via the tsetse fly, T. brucei may be transferred between mammals via bodily fluid exchange, such as by blood transfusion or sexual contact, although this is thought to be rare.[91][92] Newborn babies can be infected (vertical or congenital transmission) from infected mothers.[93]
Chemotherapy
There are four drugs generally recommended for the first-line treatment of African trypanosomiasis: suramin developed in 1921, pentamidine developed in 1941, melarsoprol developed in 1949 and eflornithine developed in 1990.[94][95] These drugs are not fully effective and are toxic to humans.[96] In addition, drug resistance has developed in the parasites against all the drugs.[97] The drugs are of limited application since they are effective against specific strains of T. brucei and the life cycle stages of the parasites. Suramin is used only for first-stage infection of T. b. rhodesiense, pentamidine for first-stage infection of T. b. gambiense, and eflornithine for second-stage infection of T. b. gambiense. Melarsopol is the only drug effective against the two types of parasite in both infection stages,[98] but is highly toxic, such that 5% of treated individuals die of brain damage (reactive encephalopathy).[99] Another drug, nifurtimox, recommended for Chagas disease (American trypanosomiasis), is itself a weak drug but in combination with melarsopol, it is used as the first-line medication against second-stage infection of T. b. gambiense.[100][101]
Historically, arsenic and mercuric compounds were introduced in the early 20th century, with success particularly in animal infections.[102][103] German physician Paul Ehrlich and his Japanese associate Kiyoshi Shiga developed the first specific trypanocidal drug in 1904 from a dye, trypan red, which they named Trypanroth.[104] These chemical preparations were effective only at high and toxic dosages, and were not suitable for clinical use.[105]
Animal trypanosomiasis is treated with six drugs: diminazene aceturate, homidium (homidium bromide and homidium chloride), isometamidium chloride, melarsomine, quinapyramine, and suramin. They are all highly toxic to animals,[106] and drug resistance is prevalent.[107] Homidium is the first prescription anti-trypanosomal drug. It was developed as a modified compound of phenantridine, which was found in 1938 to have trypanocidal activity against the bovine parasite, T. congolense.[108] Among its products, dimidium bromide and its derivatives were first used in 1948 in animal cases in Africa,[109][110][111] and became known as homidium (or as ethidium bromide in molecular biology[112]).[113][114]
Drug development
The major challenge against the human disease has been to find drugs that readily pass the blood-brain barrier. The latest drug that has come into clinical use is fexinidazol, but promising results have also been obtained with the benzoxaborole drug acoziborole (SCYX-7158). This drug is currently under evaluation as a single-dose oral treatment, which is a great advantage compared to currently used drugs. Another research field that has been extensively studied in Trypanosoma brucei is to target its nucleotide metabolism.[115] The nucleotide metabolism studies have both led to the development of adenosine analogues looking promising in animal studies, and to the finding that downregulation of the P2 adenosine transporter is a common way to acquire partial drug resistance against the melaminophenyl arsenical and diamidine drug families (containing melarsoprol and pentamidine, respectively).[115] This is particularly a problem with the veterinary drug diminazene aceturate. Drug uptake and degradation are two major issues to consider to avoid drug resistance development. In the case of nucleoside analogues, they need to be taken up by the P1 nucleoside transporter (instead of P2), and they also need to be resistant against cleavage in the parasite.[116][117]
Phytochemicals. Some phytochemicals have shown research promise against the T. b. brucei strain.[118] Aderbauer et al., 2008 and Umar et al., 2010 find Khaya senegalensis is effective in vitro and Ibrahim et al., 2013 and 2008 in vivo (in rats).[118] Ibrahim et al., 2013 find a lower dose reduces parasitemia by this subspecies and a higher dose is curative and prevents injury.[118]
Distribution
T. brucei is only found in the blue areas
T. brucei is found where its tsetse fly vectors are prevalent in continental Africa. That is to say, tropical rainforest (Af), tropical monsoon (Am), and tropical savannah (Aw) areas of continental Africa.[61] Hence, the equatorial region of Africa is called the "sleeping sickness" belt. However, the specific type of the trypanosome differs according to geography. T. b. rhodesiense is found primarily in East Africa, while T. b. gambiense is found in Central and West Africa.
Trypanosoma brucei gambiense evolved from a single progenitor ~10,000 years ago.[120] It is evolving asexually and its genome shows the Meselson effect.[120]
Genetics
There are two subpopulations of T. b. gambiense that possesses two distinct groups that differ in genotype and phenotype. Group 2 is more akin to T. b. brucei than group 1 T. b. gambiense.[121]
All T. b. gambiense are resistant to killing by a serum component — trypanosome lytic factor (TLF) of which there are two types: TLF-1 and TLF-2. Group 1 T. b. gambiense parasites avoid uptake of the TLF particles while those of group 2 are able to either neutralize or compensate for the effects of TLF.[122]
In contrast, resistance in T. b. rhodesiense is dependent upon the expression of a serum resistance associated (SRA) gene.[123] This gene is not found in T. b. gambiense.[124]
11 pairs of large chromosomes of 1 to 6 megabase pairs.
3–5 intermediate chromosomes of 200 to 500 kilobase pairs.
Around 100 minichromosomes of around 50 to 100 kilobase pairs. These may be present in multiple copies per haploid genome.
Most genes are held on the large chromosomes, with the minichromosomes carrying only VSG genes. The genome has been sequenced and is available on GeneDB.[126]
The mitochondrial genome is found condensed into the kinetoplast, an unusual feature unique to the kinetoplastid protozoans. The kinetoplast and the basal body of the flagellum are strongly associated via a cytoskeletal structure[127]
In 1993, a new base, ß-d-glucopyranosyloxymethyluracil (base J), was identified in the nuclear DNA of T. brucei.[128]
The surface of T. brucei and other species of trypanosomes is covered by a dense external coat called variant surface glycoprotein (VSG).[129] VSGs are 60-kDa proteins which are densely packed (~5 million molecules) to form a 12–15nm surface coat. VSG dimers make up about 90% of all cell surface proteins in trypanosomes. They also make up ~10% of total cell protein. For this reason, these proteins are highly immunogenic and an immune response raised against a specific VSG coat will rapidly kill trypanosomes expressing this variant. However, with each cell division there is a possibility that the progeny will switch expression to change the VSG that is being expressed.[129][130]
This VSG coat enables an infecting T. brucei population to persistently evade the host's immune system, allowing chronic infection. VSG is highly immunogenic, and an immune response raised against a specific VSG coat rapidly kills trypanosomes expressing this variant. Antibody-mediated trypanosome killing can also be observed in vitro by a complement-mediatedlysisassay. However, with each cell division there is a possibility that one or both of the progeny will switch expression to change the VSG that is being expressed. The frequency of VSG switching has been measured to be approximately 0.1% per division.[131] As T. brucei populations can peak at a size of 1011 within a host[132] this rapid rate of switching ensures that the parasite population is typically highly diverse.[133][134] Because host immunity against a specific VSG does not develop immediately, some parasites will have switched to an antigenically distinct VSG variant, and can go on to multiply and continue the infection. The clinical effect of this cycle is successive 'waves' of parasitemia (trypanosomes in the blood).[129]
Expression of VSG genes occurs through a number of mechanisms yet to be fully understood.[135] The expressed VSG can be switched either by activating a different expression site (and thus changing to express the VSG in that site), or by changing the VSG gene in the active site to a different variant. The genome contains many hundreds if not thousands of VSG genes, both on minichromosomes and in repeated sections ('arrays') in the interior of the chromosomes. These are transcriptionally silent, typically with omitted sections or premature stop codons, but are important in the evolution of new VSG genes. It is estimated up to 10% of the T. brucei genome may be made up of VSG genes or pseudogenes. It is thought that any of these genes can be moved into the active site by recombination for expression.[130] VSG silencing is largely due to the effects of histone variants H3.V and H4.V. These histones cause changes in the three-dimensional structure of the T. brucei genome that results in a lack of expression. VSG genes are typically located in the subtelomeric regions of the chromosomes, which makes it easier for them to be silenced when they are not being used.[136][137] It remains unproven whether the regulation of VSG switching is purely stochastic or whether environmental stimuli affect switching frequency. Switching is linked to two factors: variation in activation of individual VSG genes; and differentiation to the "short stumpy" stage - triggered by conditions of high population density - which is the nonreproductive, interhost transmission stage.[77]As of 2021[update] it also remains unexplained how this transition is timed and how the next surface protein gene is chosen.[2] These questions of antigenic variation in T. brucei and other parasites are among the most interesting in the field of infection.[2]
Killing by human serum and resistance to human serum killing
Trypanosoma brucei brucei (as well as related species T. equiperdum and T. evansi) is not human infective because it is susceptible to innate immune system 'trypanolytic' factors present in the serum of some primates, including humans. These trypanolytic factors have been identified as two serum complexes designated trypanolytic factors (TLF-1 and −2) both of which contain haptoglobin-related protein (HPR) and apolipoprotein LI (ApoL1). TLF-1 is a member of the high density lipoprotein family of particles while TLF-2 is a related high molecular weight serum protein binding complex.[138][139] The protein components of TLF-1 are haptoglobin related protein (HPR), apolipoprotein L-1 (apoL-1) and apolipoprotein A-1 (apoA-1). These three proteins are colocalized within spherical particles containing phospholipids and cholesterol. The protein components of TLF-2 include IgM and apolipoprotein A-I.[140]
Trypanolytic factors are found only in a few species, including humans, gorillas, mandrills, baboons and sooty mangabeys. This appears to be because haptoglobin-related protein and apolipoprotein L-1 are unique to primates. This suggests these genes originated in the primate genome 25million years ago-35million years ago.[141]
Human infective subspecies T. b. gambiense and T. b. rhodesiense have evolved mechanisms of resisting the trypanolytic factors, described below.
ApoL1
ApoL1 is a member of a six gene family, ApoL1-6, that have arisen by tandem duplication. These proteins are normally involved in host apoptosis or autophagic death and possess a Bcl-2 homology domain 3.[142]ApoL1 has been identified as the toxic component involved in trypanolysis.[143] ApoLs have been subject to recent selective evolution possibly related to resistance to pathogens.[144]
The gene encoding ApoL1 is found on the long arm of chromosome 22 (22q12.3). Variants of this gene, termed G1 and G2, provide protection against T. b. rhodesiense.[145] These benefits are not without their downside as a specific ApoL1glomerulopathy has been identified.[145][146] This glomerulopathy may help to explain the greater prevalence of hypertension in African populations.[147]
The gene encodes a protein of 383 residues, including a typical signal peptide of 12 amino acids.[148] The plasma protein is a single chain polypeptide with an apparent molecular mass of 42 kilodaltons. ApoL1 has a membrane pore forming domain functionally similar to that of bacterial colicins.[149] This domain is flanked by the membrane addressing domain and both these domains are required for parasite killing.
Hpr is 91% identical to haptoglobin (Hp), an abundant acute phase serum protein, which possesses a high affinity for hemoglobin (Hb). When Hb is released from erythrocytes undergoing intravascular hemolysis Hp forms a complex with the Hb and these are removed from circulation by the CD163 scavenger receptor. In contrast to Hp–Hb, the Hpr–Hb complex does not bind CD163 and the Hpr serum concentration appears to be unaffected by hemolysis.[152]
Killing mechanism
The association of HPR with hemoglobin allows TLF-1 binding and uptake via the trypanosome haptoglobin-hemoglobin receptor (TbHpHbR).[153] TLF-2 enters trypanosomes independently of TbHpHbR.[153] TLF-1 uptake increases when haptoglobin level is low. TLF-1 overtakes haptoglobin and binds free hemoglobin in the serum. However the complete absence of haptoglobin is associated with a decreased killing rate by serum.[154]
The trypanosome haptoglobin-hemoglobin receptor is an elongated three a-helical bundle with a small membrane distal head.[155] This protein extends above the variant surface glycoprotein layer that surrounds the parasite.
The first step in the killing mechanism is the binding of TLF to high affinity receptors—the haptoglobin-hemoglobin receptors—that are located in the flagellar pocket of the parasite.[153][156] The bound TLF is endocytosed via coated vesicles and then trafficked to the parasite lysosomes. ApoL1 is the main lethal factor in the TLFs and kills trypanosomes after insertion into endosomal / lysosomal membranes.[143] After ingestion by the parasite, the TLF-1 particle is trafficked to the lysosome wherein ApoL1 is activated by a pH mediated conformational change. After fusion with the lysosome the pH drops from ~7 to ~5. This induces a conformational change in the ApoL1 membrane addressing domain which in turn causes a salt bridge linked hinge to open. This releases ApoL1 from the HDL particle to insert in the lysosomal membrane. The ApoL1 protein then creates anionic pores in the membrane which leads to depolarization of the membrane, a continuous influx of chloride and subsequent osmotic swelling of the lysosome. This influx in its turn leads to rupture of the lysosome and the subsequent death of the parasite.[157]
Resistance mechanisms: T. b. gambiense
Trypanosoma brucei gambiense causes 97% of human cases of sleeping sickness. Resistance to ApoL1 is principally mediated by the hydrophobic β-sheet of the T. b. gambiense specific glycoprotein.[158] Other factors involved in resistance appear to be a change in the cysteine protease activity and TbHpHbR inactivation due to a leucine to serine substitution (L210S) at codon 210.[158] This is due to a thymidine to cytosine mutation at the second codon position.[159]
These mutations may have evolved due to the coexistence of malaria where this parasite is found.[158] Haptoglobin levels are low in malaria because of the hemolysis that occurs with the release of the merozoites into the blood. The rupture of the erythrocytes results in the release of free haem into the blood where it is bound by haptoglobin. The haem is then removed along with the bound haptoglobin from the blood by the reticuloendothelial system.[160]
Resistance mechanisms: T. b. rhodesiense
Trypanosoma brucei rhodesiense relies on a different mechanism of resistance: the serum resistance associated protein (SRA). The SRA gene is a truncated version of the major and variable surface antigen of the parasite, the variant surface glycoprotein.[161] However, it has little similarity (low sequence homology) with the VSG gene (<25%). SRA is an expression site associated gene in T. b. rhodesiense and is located upstream of the VSGs in the active telomeric expression site.[162] The protein is largely localized to small cytoplasmic vesicles between the flagellar pocket and the nucleus. In T. b. rhodesiense the TLF is directed to SRA containing endosomes while some dispute remains as to its presence in the lysosome.[143][163] SRA binds to ApoL1 using a coiled–coiled interaction at the ApoL1 SRA interacting domain while within the trypanosome lysosome.[143] This interaction prevents the release of the ApoL1 protein and the subsequent lysis of the lysosome and death of the parasite.
Baboons are known to be resistant to T. b. rhodesiense. The baboon version of the ApoL1 gene differs from the human gene in a number of respects including two critical lysines near the C terminus that are necessary and sufficient to prevent baboon ApoL1 binding to SRA.[164] Experimental mutations allowing ApoL1 to be protected from neutralization by SRA have been shown capable of conferring trypanolytic activity on T. b. rhodesiense.[123] These mutations resemble those found in baboons, but also resemble natural mutations conferring protection of humans against T. b. rhodesiense which are linked to kidney disease.[145]
↑Legros D, Ollivier G, Gastellu-Etchegorry M, Paquet C, Burri C, Jannin J, Büscher P (July 2002). "Treatment of human African trypanosomiasis--present situation and needs for research and development". The Lancet. Infectious Diseases. 2 (7): 437–440. doi:10.1016/S1473-3099(02)00321-3. hdl:10144/18268. PMID12127356.
↑Gibson W (July 2007). "Resolution of the species problem in African trypanosomes". International Journal for Parasitology. 37 (8–9): 829–838. doi:10.1016/j.ijpara.2007.03.002. PMID17451719.
12Joubert JJ, Schutte CH, Irons DJ, Fripp PJ (1993). "Ubombo and the site of David Bruce's discovery of Trypanosoma brucei". Transactions of the Royal Society of Tropical Medicine and Hygiene. 87 (4): 494–495. doi:10.1016/0035-9203(93)90056-v. PMID8249096.
12Cox FE (June 2004). "History of sleeping sickness (African trypanosomiasis)". Infectious Disease Clinics of North America. 18 (2): 231–245. doi:10.1016/j.idc.2004.01.004. PMID15145378.
↑Ormerod WE (October 1991). "Hypothesis: the significance of Winterbottom's sign". The Journal of Tropical Medicine and Hygiene. 94 (5): 338–340. PMID1942213.
↑Martini E (1903). "Ueber die Entwickelung der Tsetseparasiten in Säugethieren" [Preliminary note on the morphology and distribution of the parasite found in tsetse disease]. Zeitschrift für Hygiene und Infektionskrankheiten (in German). 42 (1): 341–351. doi:10.1007/BF02217469. S2CID12816407.
↑Köhler W, Köhler M (September 2002). "Zentralblatt für Bakteriologie--100 years ago: sleeping sickness--intoxication or infectious disease?". International Journal of Medical Microbiology. 292 (3–4): 141–147. doi:10.1078/1438-4221-00190. PMID12398205.
↑Cook GC (1993). "Royal Society of Tropical Medicine and Hygiene meeting at Manson House, London, 10 December 1992. George Carmichael Low FRCP: twelfth president of the Society and underrated pioneer of tropical medicine". Transactions of the Royal Society of Tropical Medicine and Hygiene. 87 (4): 355–360. doi:10.1016/0035-9203(93)90002-8. PMID8249057.
↑Castellani A (1903). "On the discovery of a species of trypanosoma in the cerebrospinal fluid of cases of sleeping sickness". The Lancet. 161 (4164): 1735–1736. doi:10.1016/S0140-6736(01)70338-8.
↑Castellani A (1903). "On the discovery of a species of trypanosoma in the cerebrospinal fluid of cases of sleeping sickness". Proceedings of the Royal Society of London. 71 (467–476): 501–508. doi:10.1098/rspl.1902.0134. S2CID59110369.
↑Bruce D, Hamerton AE, Bateman HR, Mackie FP (1909). "The development of Trypanosoma gambiense in Glossina palpalis". Proceedings of the Royal Society of London. Series B. 81 (550): 405–414. doi:10.1098/rspb.1909.0041.
↑Rifkin MR (August 1984). "Trypanosoma brucei: biochemical and morphological studies of cytotoxicity caused by normal human serum". Experimental Parasitology. 58 (1). Elsevier: 81–93. doi:10.1016/0014-4894(84)90023-7. PMID6745390.
↑Felu, Cecile; Pasture, Julie; Pays, Etienne; Pérez-Morga, David (2007). "Diagnostic potential of a conserved genomic rearrangement in the Trypanosoma brucei gambiense-specific TGSGP locus". The American Journal of Tropical Medicine and Hygiene. 76 (5): 922–929. doi:10.4269/ajtmh.2007.76.922. ISSN0002-9637. PMID17488917.
↑Bastin P, Matthews KR, Gull K (August 1996). "The paraflagellar rod of kinetoplastida: solved and unsolved questions". Parasitology Today. 12 (8): 302–307. doi:10.1016/0169-4758(96)10031-4. PMID15275181.
↑Woods A, Sherwin T, Sasse R, MacRae TH, Baines AJ, Gull K (July 1989). "Definition of individual components within the cytoskeleton of Trypanosoma brucei by a library of monoclonal antibodies". Journal of Cell Science. 93 (3): 491–500. doi:10.1242/jcs.93.3.491. PMID2606940.
12Juan T, Fürthauer M (February 2018). "Biogenesis and function of ESCRT-dependent extracellular vesicles". Seminars in Cell & Developmental Biology. 74. Elsevier: 66–77. doi:10.1016/j.semcdb.2017.08.022. PMID28807885.
↑Proto, William R.; Coombs, Graham H.; Mottram, Jeremy C. (2013). "Cell death in parasitic protozoa: regulated or incidental?". Nature Reviews. Microbiology. 11 (1): 58–66. doi:10.1038/nrmicro2929. ISSN1740-1534. PMID23202528. S2CID1633550.
↑Van Den Abbeele, J.; Claes, Y.; van Bockstaele, D.; Le Ray, D.; Coosemans, M. (1999). "Trypanosoma brucei spp. development in the tsetse fly: characterization of the post-mesocyclic stages in the foregut and proboscis". Parasitology. 118 (5): 469–478. doi:10.1017/s0031182099004217. ISSN0031-1820. PMID10363280. S2CID32217938.
↑Krisnky WL (2009). "Tsetse fly (Glossinidae)". In Mullen GR, Durden L (eds.). Medical and Veterinary Entomology (2ed.). Amsterdam: Elsevier. p.296. ISBN978-0-0-80-91969-0. Archived from the original on 1 October 2024. Retrieved 17 September 2017.
↑Nok AJ (May 2003). "Arsenicals (melarsoprol), pentamidine and suramin in the treatment of human African trypanosomiasis". Parasitology Research. 90 (1): 71–79. doi:10.1007/s00436-002-0799-9. PMID12743807. S2CID35019516.
↑Wilson SG (April 1948). "Further observations on the curative value of dimidium bromide in Trypanosoma congolense infections in bovines in Uganda". The Journal of Comparative Pathology and Therapeutics. 58 (2): 94–106. doi:10.1016/s0368-1742(48)80008-1. PMID18861668.
↑Wilson SG (1948). "Further Observations on the Curative Value of Dimidium Bromide (Phenanthridinium 1553) in Trypanosoma Congolense Infections in Bovines in Uganda". Journal of Comparative Pathology and Therapeutics. 58 (2): 94–106. doi:10.1016/S0368-1742(48)80008-1. PMID18861668.
↑Carmichael J (April 1950). "Dimidium bromide or phenanthridinium 1553; a note on the present position". The Veterinary Record. 62 (17): 257. doi:10.1136/vr.62.17.257-a (inactive 12 July 2025). PMID15418753. S2CID33016283.{{cite journal}}: CS1 maint: DOI inactive as of July 2025 (link)
↑Paindavoine P, Pays E, Laurent M, Geltmeyer Y, Le Ray D, Mehlitz D, Steinert M (February 1986). "The use of DNA hybridization and numerical taxonomy in determining relationships between Trypanosoma brucei stocks and subspecies". Parasitology. 92 (Pt 1): 31–50. doi:10.1017/S0031182000063435. PMID3960593. S2CID33529173.
↑De Greef C, Imberechts H, Matthyssens G, Van Meirvenne N, Hamers R (September 1989). "A gene expressed only in serum-resistant variants of Trypanosoma brucei rhodesiense". Molecular and Biochemical Parasitology. 36 (2): 169–176. doi:10.1016/0166-6851(89)90189-8. PMID2528066.
↑Ogbadoyi E, Ersfeld K, Robinson D, Sherwin T, Gull K (March 2000). "Architecture of the Trypanosoma brucei nucleus during interphase and mitosis". Chromosoma. 108 (8): 501–513. doi:10.1007/s004120050402. PMID10794572. S2CID3850480.
↑Turner CM (August 1997). "The rate of antigenic variation in fly-transmitted and syringe-passaged infections of Trypanosoma brucei". FEMS Microbiology Letters. 153 (1): 227–231. doi:10.1111/j.1574-6968.1997.tb10486.x. PMID9252591.
↑Pays E (November 2005). "Regulation of antigen gene expression in Trypanosoma brucei". Trends in Parasitology. 21 (11): 517–520. doi:10.1016/j.pt.2005.08.016. PMID16126458.
↑Wasser WG, Tzur S, Wolday D, Adu D, Baumstein D, Rosset S, Skorecki K (2012). "Population genetics of chronic kidney disease: the evolving story of APOL1". Journal of Nephrology. 25 (5): 603–618. doi:10.5301/jn.5000179 (inactive 12 July 2025). PMID22878977.{{cite journal}}: CS1 maint: DOI inactive as of July 2025 (link)
↑Richard Seed, John; Seed, Thomas M.; Sechelski, John (January 1978). "The biological effects of tryptophol (indole-3-ethanol): Hemolytic, biochemical and behavior modifying activity". Comparative Biochemistry and Physiology Part C: Comparative Pharmacology. 60 (2): 175–185. doi:10.1016/0306-4492(78)90091-6. PMID28889.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.