General anaesthetics are often defined as compounds that induce a loss of consciousness in humans or loss of righting reflex in animals. Clinical definitions are also extended to include an induced coma that causes lack of awareness to painful stimuli, sufficient to facilitate surgical applications in clinical and veterinary practice. General anaesthetics do not act as analgesics and should also not be confused with sedatives. General anaesthetics are a structurally diverse group of compounds whose mechanisms encompass multiple biological targets involved in the control of neuronal pathways. The precise workings are the subject of some debate and ongoing research.

Propofol is the active component of an intravenous anesthetic formulation used for induction and maintenance of general anesthesia. It is chemically termed 2,6-diisopropylphenol. The formulation was originally approved under the brand name Diprivan. Numerous generic offerings of this formulation now exist. Intravenous administration is used to induce unconsciousness after which anesthesia may be maintained using a combination of medications. It is manufactured as part of a sterile injectable emulsion formulation using soybean oil and lecithin, giving it a white milky coloration.
Absorption is the journey of a drug travelling from the site of administration to the site of action.
SCH-50911 is a selective GABAB antagonist. Its main applications are in pharmacology research.

JWH-200 (WIN 55,225) is an analgesic chemical from the aminoalkylindole family that acts as a cannabinoid receptor agonist. Its binding affinity, Ki at the CB1 receptor is 42 nM, around the same as that of THC, but its analgesic potency in vivo was higher than that of other analogues with stronger CB1 binding affinity in vitro, around 3 times that of THC but with less sedative effect, most likely reflecting favourable pharmacokinetic characteristics. It was discovered in 1991 by Sterling Drug as a potential analgesic following the earlier identification of related compounds such as pravadoline and WIN 55,212-2.
MS-245 is a tryptamine derivative used in scientific research. It acts as a selective 5-HT6 receptor antagonist with a Ki of 2.3 nM, and was derived through structure-activity relationship development of the selective 5-HT6 agonist EMDT. It has been used as a lead compound for further development of tryptamine-derived 5-HT6 antagonists. In animal studies it has been shown to boost the activity of, but not substitute for, both amphetamine and nicotine.
ORG-20599 is a synthetic neuroactive steroid, with sedative effects resulting from its action as a GABAA receptor positive allosteric modulator and, at higher concentrations, agonist. It was developed for use as an anaesthetic agent but was never marketed for this purpose, although it is still used in scientific research.

L-371,257 is a compound used in scientific research which acts as a selective antagonist of the oxytocin receptor with over 800x selectivity over the related vasopressin receptors. It was one of the first non-peptide oxytocin antagonists developed, and has good oral bioavailability, but poor penetration of the blood–brain barrier, which gives it good peripheral selectivity with few central side effects. Potential applications are likely to be in the treatment of premature labour.

JWH-007 is an analgesic chemical from the naphthoylindole family, which acts as a cannabinoid agonist at both the CB1 and CB2 receptors. It was first reported in 1994 by a group including the noted cannabinoid chemist John W. Huffman. It was the most active of the first group of N-alkyl naphoylindoles discovered by the team led by John W Huffman, several years after the family was initially described with the discovery of the N-morpholinylethyl compounds pravadoline (WIN 48,098), JWH-200 (WIN 55,225) and WIN 55,212-2 by the Sterling Winthrop group. Several other N-alkyl substituents were found to be active by Huffman's team including the n-butyl, n-hexyl, 2-heptyl, and cyclohexylethyl groups, but it was subsequently determined that the 2-methyl group on the indole ring is not required for CB1 binding, and tends to increase affinity for CB2 instead. Consequently, the 2-desmethyl derivative of JWH-007, JWH-018, has slightly higher binding affinity for CB1, with an optimum binding of 9.00 nM at CB1 and 2.94 nM at CB2, and JWH-007 displayed optimum binding of 9.50 nM at CB1 and 2.94 nM at CB2.
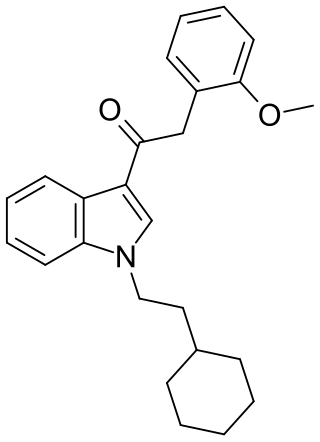
RCS-8 (also known as 1-(2-cyclohexylethyl)-3-(2-methoxyphenylacetyl)indole, SR-18, and BTM-8) is a synthetic cannabinoid that has been found as an ingredient of "herbal" synthetic cannabis blends. It can be described as an analogue of JWH-250 with the 1-pentyl group replaced by 1-(2-cyclohexylethyl), and can be expected to be less potent than JWH-250 (cf. JWH-007 and its cyclohexylethyl analogue). Despite not having been reported in the scientific or patent literature as yet, reputed recreational use of RCS-8 in the United States has led to it being specifically listed in a proposed 2011 amendment to the Controlled Substances Act, aiming to add a number of synthetic drugs into Schedule I. In addition, all CB1 receptor agonists of the 3-phenylacetylindole class such as RCS-8 are Schedule I Controlled Substances.

Elinogrel (INN, USAN) was an experimental antiplatelet drug acting as a P2Y12 inhibitor. Similarly to ticagrelor and in contrast to clopidogrel, elinogrel was a reversible inhibitor that acted fast and short (for about 12 hours), and it was not a prodrug but pharmacologically active itself. The substance was used in form of its potassium salt, intravenously for acute treatment and orally for long-term treatment. Development was terminated in 2012.
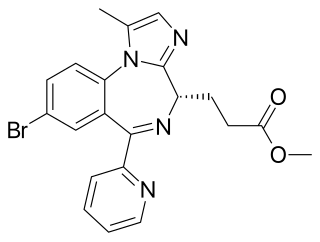
Remimazolam, sold under the brand name Byfavo, is a medication for the induction and maintenance of procedural sedation in adults for invasive diagnostic or surgical procedures lasting 30 minutes or less. It is a benzodiazepine drug, developed by PAION AG in collaboration with several regional licensees as an alternative to the short-acting imidazobenzodiazepine midazolam, for use in the induction of anesthesia and conscious sedation for minor invasive procedures. Remimazolam was found to have both a more rapid onset and a shorter duration than midazolam, and human clinical trials showed a faster recovery time and predictable, consistent pharmacokinetics, suggesting some advantages over existing drugs for these applications.
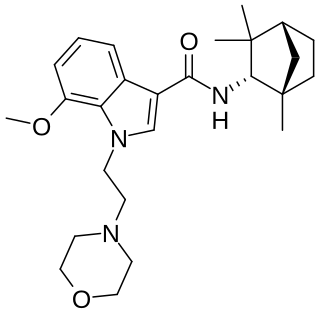
MN-25 (UR-12) is a drug invented by Bristol-Myers Squibb, that acts as a reasonably selective agonist of peripheral cannabinoid receptors. It has moderate affinity for CB2 receptors with a Ki of 11 nM, but 22x lower affinity for the psychoactive CB1 receptors with a Ki of 245 nM. The indole 2-methyl derivative has the ratio of affinities reversed however, with a Ki of 8 nM at CB1 and 29 nM at CB2, which contrasts with the usual trend of 2-methyl derivatives having increased selectivity for CB2 (cf. JWH-018 vs JWH-007, JWH-081 vs JWH-098).
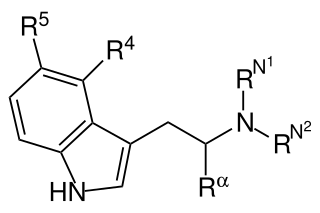
Substituted tryptamines, or serotonin analogues, are organic compounds which may be thought of as being derived from tryptamine itself. The molecular structures of all tryptamines contain an indole ring, joined to an amino (NH2) group via an ethyl (−CH2–CH2−) sidechain. In substituted tryptamines, the indole ring, sidechain, and/or amino group are modified by substituting another group for one of the hydrogen (H) atoms.

ORG-25435 is a synthetic drug developed by Organon International, which acts as a GABAA receptor positive allosteric modulator, and produces sedative effects. It has been researched for use as an intravenous anesthetic agent, with positive results in initial trials, although negative side effects like hypotension and tachycardia, as well as unpredictable pharmacokinetics at higher doses, have meant it has ultimately not been adopted for medical use.

RU-58841, also known as PSK-3841 or HMR-3841, is a nonsteroidal antiandrogen (NSAA) which was initially developed in the 1980s by Roussel Uclaf, the French pharmaceutical company from which it received its name. It was formerly under investigation by ProStrakan for potential use as a topical treatment for androgen-dependent conditions including acne, pattern hair loss, and excessive hair growth. The compound is similar in structure to the NSAA RU-58642 but contains a different side-chain. These compounds are similar in chemical structure to nilutamide, which is related to flutamide, bicalutamide, and enzalutamide, all of which are NSAAs similarly. RU-58841 can be synthesized either by building the hydantoin moiety or by aryl coupling to 5,5-dimethylhydantoin.

Hydroxybupropion, or 6-hydroxybupropion, is the major active metabolite of the antidepressant and smoking cessation drug bupropion. It is formed from bupropion by the liver enzyme CYP2B6 during first-pass metabolism. With oral bupropion treatment, hydroxybupropion is present in plasma at area under the curve concentrations that are as many as 16–20 times greater than those of bupropion itself, demonstrating extensive conversion of bupropion into hydroxybupropion in humans. As such, hydroxybupropion is likely to play a very important role in the effects of oral bupropion, which could accurately be thought of as functioning largely as a prodrug to hydroxybupropion. Other metabolites of bupropion besides hydroxybupropion include threohydrobupropion and erythrohydrobupropion.

The ProTide technology is a prodrug approach used in molecular biology and drug design. It is designed to deliver nucleotide analogues into the cell. This technology was invented by Professor Chris McGuigan from the School of Pharmacy and Pharmaceutical Sciences at Cardiff University in the early 1990s. ProTides form a critical part of antiviral drugs such as sofosbuvir, tenofovir alafenamide, and remdesivir.
Total intravenous anesthesia (TIVA) refers to the intravenous administration of anesthetic agents to induce a temporary loss of sensation or awareness. The first study of TIVA was done in 1872 using chloral hydrate, and the common anesthetic agent propofol was licensed in 1986. TIVA is currently employed in various procedures as an alternative technique of general anesthesia in order to improve post-operative recovery.

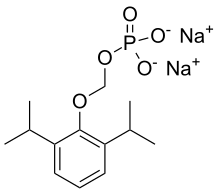
Ciprofol is a novel 2,6-disubstituted phenol derivative that is used for the intravenous induction of general anesthesia. A short-acting and highly selective γ-aminobutyric acid agonist, ciprofol is 4–6 times more potent than other phenol derivatives such as propofol or fospropofol.