The human Y chromosome showing the SRY gene which codes for a protein regulating sexual differentiation.
Sexual differentiation in humans is the process of development of sex differences in humans. It is defined as the development of phenotypic structures consequent to the action of hormones produced following gonadal determination.[1]Sexual differentiation includes development of different genitals and internal genital tracts; body hair also plays a role in sex identification.[2]
The development of sexual differences begins with the XY sex-determination system that is present in humans, and complex mechanisms are responsible for the development of the phenotypic differences between male and femalehumans from an undifferentiated zygote.[3] Females typically have two X chromosomes, and males typically have a Y chromosome and an X chromosome. At an early stage in embryonic development, both sexes possess equivalent internal structures. These are the mesonephric ducts and paramesonephric ducts. The presence of the SRY gene on the Y chromosome causes the development of the testes in males, and the subsequent release of hormones which cause the paramesonephric ducts to regress. In females, the mesonephric ducts regress.
Disorders of sexual development (DSD), encompassing conditions characterized by the appearance of undeveloped genitals that may be ambiguous, or look like those typical for the opposite sex, sometimes known as intersex, can be a result of genetic and hormonal factors.[4]
Most mammals, including humans, have an XY sex-determination system: the Y chromosome carries factors responsible for triggering male development. In the absence of a Y chromosome, the fetus will undergo female development. This is because of the presence of the sex-determining region of the Y chromosome, also known as the SRY gene.[5] Thus, male mammals typically have an X and a Y chromosome (XY), while female mammals typically have two X chromosomes (XX).
Chromosomal sex is determined at the time of fertilization; a chromosome from the sperm cell, either X or Y, fuses with the X chromosome in the egg cell. Gonadal sex refers to the gonads, that is the testicles or ovaries, depending on which genes are expressed. Phenotypic sex refers to the structures of the external and internal genitalia.[6][7][8][9]
Six weeks elapse after fertilization before the first signs of sex differentiation can be observed in human embryos.[5] The embryo and subsequent early fetus appear to be sexually indifferent, looking neither like a male or a female. Over the next several weeks, hormones are produced that cause undifferentiated tissue to transform into either male or female reproductive organs. This process is called sexual differentiation. The precursor of the internal female sex organs is called the Müllerian system.
Differentiation between the sexes of the sex organs occurs throughout embryological, fetal and later life. In both males and females, the sex organs consist of two structures: the internal genitalia and the external genitalia. In males, the gonads are the testicles and in females, they are the ovaries. These are the organs that produce gametes (egg and sperm), the reproductive cells that will eventually meet to form the fertilized egg (zygote).
As the zygote divides, it first becomes the embryo (which means 'growing within'), typically between zero and eight weeks, then from the eighth week until birth, it is considered the fetus (which means 'unborn offspring'). The internal genitalia are all the accessory glands and ducts that connect the gonads to the outside environment. The external genitalia consist of all the external reproductive structures. The sex of an early embryo cannot be determined because the reproductive structures do not differentiate until the seventh week. Prior to this, the child is considered bipotential because it cannot be identified as male or female.
Internal genital differentiation
The internal genitalia consist of two accessory ducts: mesonephric ducts (Woffian duct) and paramesonephric ducts (Müllerian ducts). The mesonephric system is the precursor to the male genitalia and the paramesonephric to the female reproductive system.[11] As development proceeds, one of the pairs of ducts develops while the other regresses. This depends on the presence or absence of the sex determining region of the Y chromosome, also known as the SRY gene.[5] In the presence of a functional SRY gene, the bipotential gonads develop into testes. Gonads are histologically distinguishable by 6–8 weeks of gestation.
Subsequent development of one set and degeneration of the other depends on the presence or absence of two testicular hormones: testosterone and anti-Müllerian hormone (AMH). Disruption of typical development may result in the development of both, or neither, duct system, which may produce morphologically intersex individuals.
Males: The SRY gene when transcribed and processed produces SRY protein that binds to DNA and directs the development of the gonad into testes. Male development can only occur when the fetal testis secretes key hormones at a critical period in early gestation. The testes begin to secrete three hormones that influence the male internal and external genitalia: they secrete anti-Müllerian hormone (AMH), testosterone, and dihydrotestosterone (DHT). Anti-Müllerian hormone causes the paramesonephric ducts to regress. Testosterone converts the mesonephric ducts into male accessory structures, including the epididymides, vasa deferentia, and seminal vesicles. Testosterone will also control the descending of the testes from the abdomen.[1] Many other genes found on other autosomes, including WT1, SOX9 and SF1 also play a role in gonadal development.[12]
Females: Without testosterone and AMH, the mesonephric ducts degenerate and disappear. The paramesonephric ducts develop into the uterus, fallopian tubes, and upper vagina (the lower vagina develops from the urogenital sinus).[12] There still remains a broad lack of information about the genetic controls of female development (as of 1992), and much remains unknown about the female embryonic process.[13] The mesonephric ducts are not completely useless in the female case: they secrete WNT9B, which is necessary for the elongation of the paramesonephric ducts. Elongation also happens through the active migration of the paramesonephric epithelium, which happens through a phosphoinositide 3-kinase pathway.[14]
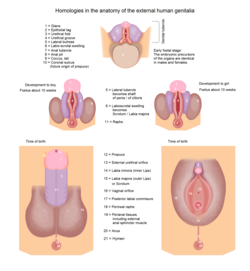
External genital differentiation
Development of external genitalia
By 7 weeks, a fetus has a genital tubercle, urogenital sinus, urogenital folds and labioscrotal swellings. In females, without excess androgens, these become the vulva (clitoris, vestibule, labia minora and labia majora respectively). Males become externally distinct between 8 and 12 weeks, as androgens enlarge the genital tubercle and cause the urogenital groove and sinus to fuse in the midline, producing an unambiguous penis with a phallic urethra, and the labioscrotal swellings become a thinned, rugate scrotum where the testicles are situated. Dihydrotestosterone will differentiate the remaining male characteristics of the external genitalia.[1]
A sufficient amount of any androgen can cause external masculinization. The most potent is dihydrotestosterone (DHT), generated from testosterone in skin and genital tissue by the action of 5α-reductase. A male fetus may be incompletely masculinized if this enzyme is deficient. In some diseases and circumstances, other androgens may be present in high enough concentrations to cause partial or (rarely) complete masculinization of the external genitalia of a genetically female fetus. The testes begin to secrete three hormones that influence the male internal and external genitalia. They secrete anti-Müllerian hormone, testosterone, and Dihydrotestosterone. Anti-Müllerian hormone (AMH) causes the paramesonephric ducts to regress. Testosterone, which is secreted and converts the mesonephric ducts into male accessory structures, such as epididymis, vas deferens and seminal vesicle. Testosterone will also control the descending of the testes from the abdomen into the scrotum. Dihydrotestosterone, also known as (DHT) will differentiate the remaining male characteristics of the external genitalia.[15]
Further sex differentiation of the external genitalia occurs at puberty, when androgen levels again become disparate. Male levels of testosterone directly induce growth of the penis, and indirectly (via DHT) the prostate.
Alfred Jost observed that while testosterone was required for mesonephric duct development, the regression of the paramesonephric duct was due to another substance. This was later determined to be paramesonephric inhibiting substance (MIS), a 140 kD dimeric glycoprotein that is produced by Sertoli cells. MIS blocks the development of paramesonephric ducts, promoting their regression.[16] Today it’s better known as the anti-Müllerian hormone (AMH).
This section needs expansion. You can help by adding to it. (January 2020)
Breast development
Visible differentiation occurs at puberty, when estradiol and other hormones cause breasts to develop in typical females.
Psychological and behavioral differentiation
Human adults and children show many psychological and behavioral sex differences. Some (e.g. dress) are learned and cultural. Others are demonstrable across cultures and have both biological and learned determinants. For example, some studies claim girls are, on average, more verbally fluent than boys, but boys are, on average, better at spatial calculation.[17] It seems likely that this is due to males generally having a greater area allocated to the space-specialized parietal cortex, while females generally have relatively more brain area allocated to the verbal-associative-specialized temporal cortex.[18]
Disorders of sex determination (DSD) are classified into a multitude of categories.[19] These categories consists of different types of female disorders along with categories specifically for male DSDs. There are also sex chromosomal DSDs such as, the later mentioned, Klinefelter and Turner syndrome[19]
The following are some of the conditions associated with atypical determination and differentiation process:[20]
A zygote with only X chromosome (XO) results in Turner syndrome and will develop with female characteristics.[5]
Congenital adrenal hyperplasia –Inability of adrenal to produce sufficient cortisol, leading to increased production of testosterone resulting in severe masculinization of 46 XX females. The condition also occurs in XY males, as they suffer from the effects of low cortisol and salt-wasting, not virilization.
Persistent Müllerian duct syndrome – A rare type of pseudohermaphroditism that occurs in 46 XY males, caused by either a mutation in the Müllerian inhibiting substance (MIS) gene, on 19p13, or its type II receptor, 12q13. Results in a retention of Müllerian ducts (persistence of rudimentary uterus and fallopian tubes in otherwise normally virilized males), unilateral or bilateral undescended testes, and sometimes causes infertility.
XY differences of sex development – Atypical androgen production or inadequate androgen response, which can cause incomplete masculinization in XY males. Varies from mild failure of masculinization with undescended testes to complete sex reversal and female phenotype (Androgen insensitivity syndrome)
Swyer syndrome. A form of complete gonadal dysgenesis, mostly due to mutations in the first step of sex determination; the SRY genes.
A 5-alpha-reductase deficiency results in atypical development characterized by female phenotype or under virilized male phenotype with development of the epididymis, vas deferens, seminal vesicle, and ejaculatory duct, but also a pseudovagina. This is because testosterone is converted to the more potent DHT by 5-alpha reductase. DHT is necessary to exert androgenic effects farther from the site of testosterone production, where the concentrations of testosterone are too low to have any potency.
Klinefelter syndrome (47,XXY)- A chromosomal disorder that results in an extra X chromosome in males. This leads to hormonal problems later on. However, this disorder while in some cases easily identified, sometimes is not extreme and can not be determined until after puberty if at all.[21]
↑Sizonenko, P. C. (n.d.). "Human sexual differentiation". Reproductive health– via Geneva Foundation for Medical Education and Research.
↑Mukherjee, Asit B.; Parsa, Nasser Z. (1990). "Determination of sex chromosomal constitution and chromosomal origin of drumsticks, drumstick-like structures, and other nuclear bodies in human blood cells at interphase by fluorescence in situ hybridization". Chromosoma. 99 (6): 432–435. doi:10.1007/BF01726695. PMID2176962. S2CID25732504.
1234Rey, Rodolfo; Josso, Nathalie; Racine, Chrystèle (27 May 2020) [first published 2000]. "Sexual Differentiation". In Feingold, Kenneth R.; Anawalt, Bradley; Blackman Marc R.; etal. (eds.). Endotext [Internet]. South Dartmouth, Mass.: MDText.com, Inc. PMID25905232. Retrieved 28 March 2023– via National Institutes of Health.
↑Achermann, John; Jameson, Larry (2012). Fauci, Anthony S. (ed.). Harrison's principles of internal medicine (18thed.). New York: McGraw-Hill Medical. pp.3046–3048. ISBN978-0-07-147693-5.
↑Purves, Dale; Augustine, George J.; Fitzpatrick, David; Katz, Lawrence C.; LaMantia, Anthony-Samuel; McNamara, James O.; Williams, S. Mark (2001), "What Is Sex?", Neuroscience. 2nd edition, Sinauer Associates, retrieved 27 December 2025
↑Silverthorn, Dee, U.. (2010). Reproduction and Development. In: Human Physiology: an integrated approach. 5th ed. san francisco: Pearson education. pp. 828–831.
↑Fausto-Sterling, Anne (1992). Myths Of Gender: Biological Theories About Women And Men (reviseded.). New York: Basic Books. pp.81–82. ISBN978-0-4650-4792-5.
↑Hänggi, Jürgen (2010). "Sexual Dimorphism in the Parietal Substrate Associated with Visuospatial Cognition Independent of General Intelligence". Journal of Cognitive Neuroscience. 22 (1): 139–155. doi:10.1162/jocn.2008.21175. PMID19199407.[pageneeded]
Sharman, GB; Hughes, RL; Cooper, DW (1989). "The Chromosomal Basis of Sex-Differentiation in Marsupials". Australian Journal of Zoology. 37 (3): 451. doi:10.1071/ZO9890451.
Watson, CM; Margan, SH; Johnston, PG (1998). "Sex-chromosome elimination in the bandicoot Isoodon macrourus using Y-linked markers". Cytogenetics and Cell Genetics. 81 (1): 54–59. doi:10.1159/000015008. PMID9691176. S2CID20042866.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.