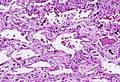
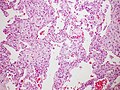
Acute interstitial pneumonitis (also known as acute interstitial pneumonia) is a rare, severe lung disease that usually affects otherwise healthy individuals.
Acute interstitial pneumonitis is often categorized as both an interstitial lung disease and a form of acute respiratory distress syndrome (ARDS). In uncommon instances, if ARDS appears acutely, in the absence of known triggers, and follows a rapidly progressing clinical course, the term "Acute interstitial pneumonia" is used.[1] ARDS is distinguished from the chronic forms of interstitial pneumonia such as idiopathic pulmonary fibrosis.[2] There is no proved treatment and management is largely based on supportive care.[3]
Symptoms and signs
The most common symptoms of acute interstitial pneumonitis are highly productive cough with expectoration of thick mucus, fever, and difficulties breathing. These often occur over a period of one to two weeks before medical attention is sought. The presence of fluid means the person experiences a feeling similar to 'drowning'. Difficulties breathing can quickly progress to an inability to breathe without support (respiratory failure).[citation needed]
Acute interstitial pneumonitis typically progresses rapidly, with hospitalization and mechanical ventilation often required only days to weeks after initial symptoms of cough, fever, and difficulties breathing develop.[3] Additional symptoms that may occur before disease onset include myalgias, fatigue, and chills.[4] Also, tachypnea and crackling noises are commonly heard when listening to the lungs of patients with this disease.
Diagnosis
Rapid progression from initial symptoms to respiratory failure is a key feature. An X-ray that shows ARDS is necessary for diagnosis (fluid in the small air sacs (alveoli) in both lungs). In addition, a biopsy of the lung that shows organizing diffuse alveolar damage is required for diagnosis. This type of alveolar damage can be attributed to nonconcentrated and nonlocalized alveoli damage, marked alveolar septal edema with inflammatory cell infiltration, fibroblast proliferation, occasional hyaline membranes, and thickening of the alveolar walls. The septa are lined with atypical, hyperplastic type II pneumocytes, thus leading to the collapse of airspaces. Other diagnostic tests are useful in excluding other similar conditions, but history, X-ray, and biopsy are essential. These other tests may include basic blood work, blood cultures, and bronchoalveolar lavage.[3]
The clinical picture is similar to ARDS, but AIP differs from ARDS in that the cause for AIP is not known.
Acute interstitial pneumonia showing a marked reduction in lung capacity
Treatment
Treatment is primarily supportive. Management in an intensive care unit is required and the need for mechanical ventilation is common. Therapy with corticosteroids is generally attempted, though their usefulness has not been established. Lung transplantation has been shown to improve survival and quality of life in selected patients with advanced or end-stage interstitial lung disease, including cases of acute interstitial pneumonitis, with median post-transplant survival reported at around five years.[5]
Prognosis
Sixty percent of people with acute interstitial pneumonitis will die in the first six months of illness.[6] The median survival is 1+1⁄2 months. However, many people who recover from the initial course of the disease experience recurrent episodes of the disease or develop chronic progressive interstitial lung disease. Although some studies have observed partial or (very rarely) complete recovery of lung function in some survivors of acute interstitial pneumonitis, the majority of survivors will continue to endure progressive lung complications.[3]
Epidemiology
Acute interstitial pneumonitis occurs most frequently among people older than forty years old. It affects men and women equally. There are no known risk factors; but smoking is implicated in the clinical behaviour of the disease.[7]
History
Acute interstitial pneumonitis was first described in 1935 by Louis Hamman and Arnold Rich, and given the name Hamman–Rich syndrome.[8]
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.