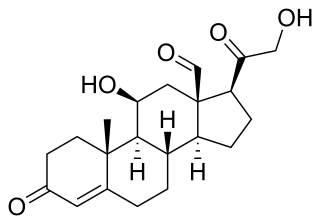
Aldosterone is the main mineralocorticoid steroid hormone produced by the zona glomerulosa of the adrenal cortex in the adrenal gland. It is essential for sodium conservation in the kidney, salivary glands, sweat glands, and colon. It plays a central role in the homeostatic regulation of blood pressure, plasma sodium (Na+), and potassium (K+) levels. It does so primarily by acting on the mineralocorticoid receptors in the distal tubules and collecting ducts of the nephron. It influences the reabsorption of sodium and excretion of potassium (from and into the tubular fluids, respectively) of the kidney, thereby indirectly influencing water retention or loss, blood pressure, and blood volume. When dysregulated, aldosterone is pathogenic and contributes to the development and progression of cardiovascular and kidney disease. Aldosterone has exactly the opposite function of the atrial natriuretic hormone secreted by the heart.
Primary aldosteronism (PA), also known as primary hyperaldosteronism or Conn's syndrome, refers to the excess production of the hormone aldosterone from the adrenal glands, resulting in low renin levels and high blood pressure. This abnormality is caused by hyperplasia or tumors. Many experience fatigue, potassium deficiency and high blood pressure which may cause poor vision, confusion or headaches. Symptoms may also include: muscular aches and weakness, muscle spasms, low back and flank pain from the kidneys, trembling, tingling sensations, dizziness/vertigo, nocturia and excessive urination. Complications include cardiovascular disease such as stroke, myocardial infarction, kidney failure and abnormal heart rhythms.
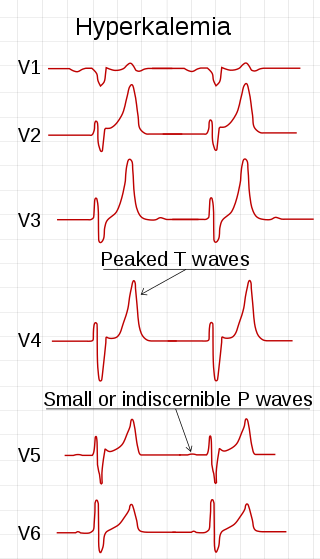
Hyperkalemia is an elevated level of potassium (K+) in the blood. Normal potassium levels are between 3.5 and 5.0 mmol/L (3.5 and 5.0 mEq/L) with levels above 5.5 mmol/L defined as hyperkalemia. Typically hyperkalemia does not cause symptoms. Occasionally when severe it can cause palpitations, muscle pain, muscle weakness, or numbness. Hyperkalemia can cause an abnormal heart rhythm which can result in cardiac arrest and death.

Loop diuretics are diuretics that act on the Na-K-Cl cotransporter along the thick ascending limb of the loop of Henle in nephrons of the kidneys. They are primarily used in medicine to treat hypertension and edema often due to congestive heart failure or chronic kidney disease. While thiazide diuretics are more effective in patients with normal kidney function, loop diuretics are more effective in patients with impaired kidney function.

Amiloride, sold under the trade name Midamor among others, is a medication typically used with other medications to treat high blood pressure or swelling due to heart failure or cirrhosis of the liver. Amiloride is classified as a potassium-sparing diuretic. Amiloride is often used together with another diuretic, such as a thiazide or loop diuretic. It is taken by mouth. Onset of action is about two hours and it lasts for about a day.
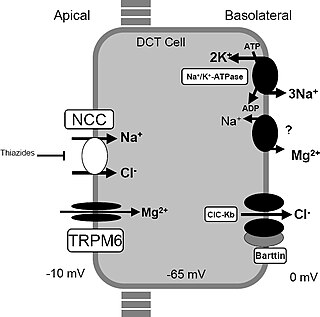
Gitelman syndrome (GS) is an autosomal recessive kidney tubule disorder characterized by low blood levels of potassium and magnesium, decreased excretion of calcium in the urine, and elevated blood pH. It is the most frequent hereditary salt-losing tubulopathy. Gitelman syndrome is caused by disease-causing variants on both alleles of the SLC12A3 gene. The SLC12A3 gene encodes the thiazide-sensitive sodium-chloride cotransporter, which can be found in the distal convoluted tubule of the kidney.

Potassium-sparing diuretics refers to drugs that cause diuresis without causing potassium loss in the urine. They are typically used as an adjunct in management of hypertension, cirrhosis, and congestive heart failure. The steroidal aldosterone antagonists can also be used for treatment of primary hyperaldosteronism. Spironolactone, a steroidal aldosterone antagonist, is also used in management of female hirsutism and acne from PCOS or other causes.
Hypoaldosteronism is an endocrinological disorder characterized by decreased levels of the hormone aldosterone. Similarly, isolated hypoaldosteronism is the condition of having lowered aldosterone without corresponding changes in cortisol.

Metabolic alkalosis is a metabolic condition in which the pH of tissue is elevated beyond the normal range (7.35–7.45). This is the result of decreased hydrogen ion concentration, leading to increased bicarbonate, or alternatively a direct result of increased bicarbonate concentrations. The condition typically cannot last long if the kidneys are functioning properly.
Hyperaldosteronism is a medical condition wherein too much aldosterone is produced by the adrenal glands, which can lead to lowered levels of potassium in the blood (hypokalemia) and increased hydrogen ion excretion (alkalosis).

Apparent mineralocorticoid excess is an autosomal recessive disorder causing hypertension, hypernatremia and hypokalemia. It results from mutations in the HSD11B2 gene, which encodes the kidney isozyme of 11β-hydroxysteroid dehydrogenase type 2. In an unaffected individual, this isozyme inactivates circulating cortisol to the less active metabolite cortisone. The inactivating mutation leads to elevated local concentrations of cortisol in the aldosterone sensitive tissues like the kidney. Cortisol at high concentrations can cross-react and activate the mineralocorticoid receptor due to the non-selectivity of the receptor, leading to aldosterone-like effects in the kidney. This is what causes the hypokalemia, hypertension, and hypernatremia associated with the syndrome. Patients often present with severe hypertension and end-organ changes associated with it like left ventricular hypertrophy, retinal, renal and neurological vascular changes along with growth retardation and failure to thrive. In serum both aldosterone and renin levels are low.
An antimineralocorticoid, also known as a mineralocorticoid receptor antagonist or aldosterone antagonist, is a diuretic drug which antagonizes the action of aldosterone at mineralocorticoid receptors. This group of drugs is often used as adjunctive therapy, in combination with other drugs, for the management of chronic heart failure. Spironolactone, the first member of the class, is also used in the management of hyperaldosteronism and female hirsutism. Most antimineralocorticoids, including spironolactone, are steroidal spirolactones. Finerenone is a nonsteroidal antimineralocorticoid.

Bartter syndrome (BS) is a rare inherited disease characterised by a defect in the thick ascending limb of the loop of Henle, which results in low potassium levels (hypokalemia), increased blood pH (alkalosis), and normal to low blood pressure. There are two types of Bartter syndrome: neonatal and classic. A closely associated disorder, Gitelman syndrome, is milder than both subtypes of Bartter syndrome.
Pseudohyperaldosteronism is a medical condition which mimics the effects of elevated aldosterone (hyperaldosteronism) by presenting with high blood pressure (hypertension), low blood potassium levels (hypokalemia), metabolic alkalosis, and low levels of plasma renin activity (PRA). However, unlike hyperaldosteronism, this conditions exhibits low or normal levels of aldosterone in the blood. Causes include genetic disorders, acquired conditions, metabolic disorders, and dietary imbalances including excessive consumption of licorice. Confirmatory diagnosis depends on the specific root cause and may involve blood tests, urine tests, or genetic testing; however, all forms of this condition exhibit abnormally low concentrations of both plasma renin activity (PRA) and plasma aldosterone concentration (PAC) which differentiates this group of conditions from other forms of secondary hypertension. Treatment is tailored to the specific cause and focuses on symptom control, blood pressure management, and avoidance of triggers.
Pseudohypoaldosteronism (PHA) is a condition that mimics hypoaldosteronism. However, the condition is due to a failure of response to aldosterone, and levels of aldosterone are actually elevated, due to a lack of feedback inhibition.

The SCNN1B gene encodes for the β subunit of the epithelial sodium channel ENaC in vertebrates. ENaC is assembled as a heterotrimer composed of three homologous subunits α, β, and γ or δ, β, and γ. The other ENAC subunits are encoded by SCNN1A, SCNN1G, and SCNN1D.
The SCNN1G gene encodes for the γ subunit of the epithelial sodium channel ENaC in vertebrates. ENaC is assembled as a heterotrimer composed of three homologous subunits α, β, and γ or δ, β, and γ. The other ENAC subunits are encoded by SCNN1A, SCNN1B, and SCNN1D.
In physiology, aldosterone escape is a term that has been used to refer to two distinct phenomena involving aldosterone that are exactly opposite each other:
- Escape from the sodium-retaining effects of excess aldosterone in primary hyperaldosteronism, manifested by volume and/or pressure natriuresis.
- The inability of ACE inhibitor therapy to reliably suppress aldosterone release, for example, in patients with heart failure or diabetes, usually manifested by increased salt and water retention. This latter sense may rather be termed refractory hyperaldosteronism.
Feline hyperaldosteronism is a disease in cats. The symptoms are caused by abnormally high concentrations of the hormone aldosterone, which is secreted by the adrenal gland. The high concentrations of aldosterone may be due directly to a disorder of the adrenal gland, or due to something outside of the adrenal gland causing it to secrete excessive aldosterone.

A diuretic is any substance that promotes diuresis, the increased production of urine. This includes forced diuresis. A diuretic tablet is sometimes colloquially called a water tablet. There are several categories of diuretics. All diuretics increase the excretion of water from the body, through the kidneys. There exist several classes of diuretic, and each works in a distinct way. Alternatively, an antidiuretic, such as vasopressin, is an agent or drug which reduces the excretion of water in urine.