



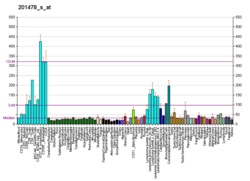
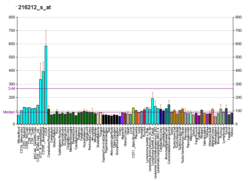
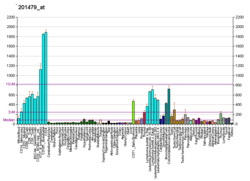
H/ACA ribonucleoprotein complex subunit 4 is a protein that in humans is encoded by the gene DKC1. [5] [6] [7]
Dyskerin is a pseudouridine synthase enzyme which is part of the TruB family of enzymes. [8] Dyskerin is an L-shaped protein of 514 residues and a molecular weight of about 58 kilo- daltons. [8] Dyskerin is essential for the activity of telomerase by accumulating telomerase RNA component (TERC). [8]
This gene is a member of the H/ACA snoRNPs (small nucleolar ribonucleoproteins) gene family. snoRNPs are involved in various aspects of rRNA processing and modification and have been classified into two families: C/D and H/ACA. The H/ACA snoRNPs also include the NOLA1, 2 and 3 proteins. The protein encoded by this gene and the three NOLA proteins localize to the dense fibrillar components of nucleoli and to coiled (Cajal) bodies in the nucleus. Both 18S rRNA production and rRNA pseudouridylation are impaired if any one of the four proteins is depleted. The protein encoded by this gene is related to the Saccharomyces cerevisiae Cbf5p and Drosophila melanogaster Nop60B proteins. The gene lies in a tail-to-tail orientation with the palmitoylated erythrocyte membrane protein (MPP1) gene and is transcribed in a telomere to centromere direction. Both nucleotide substitutions and single trinucleotide repeat polymorphisms have been found in this gene. Mutations in this gene cause X-linked dyskeratosis congenita. [7]