| Leber's congenital amaurosis | |
|---|---|
| Specialty | Ophthalmology |
| Symptoms | Visual impairment, sensitivity to light [1] |
| Types | > 12 types [1] |
| Causes | Genetic (autosomal recessive) [1] |
| Frequency | 1 in 40,000 newborns [1] |
Leber congenital amaurosis (LCA) is a rare inherited eye disease that appears at birth or in the first few months of life. [2]

Heredity, also called inheritance or biological inheritance, is the passing on of traits from parents to their offspring; either through asexual reproduction or sexual reproduction, the offspring cells or organisms acquire the genetic information of their parents. Through heredity, variations between individuals can accumulate and cause species to evolve by natural selection. The study of heredity in biology is genetics.
Contents
- Signs and symptoms
- Genetics
- Diagnosis
- Treatment
- Popular culture
- See also
- References
- Further reading
- External links
One form of LCA was successfully treated with gene therapy in 2008. [3] [4] [5] [6]
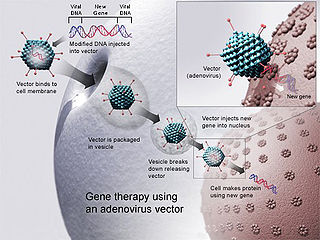
In the medicine field gene therapy is the therapeutic delivery of nucleic acid into a patient's cells as a drug to treat disease. The first attempt at modifying human DNA was performed in 1980 by Martin Cline, but the first successful nuclear gene transfer in humans, approved by the National Institutes of Health, was performed in May 1989. The first therapeutic use of gene transfer as well as the first direct insertion of human DNA into the nuclear genome was performed by French Anderson in a trial starting in September 1990.
It affects about 1 in 40,000 newborns. [1] LCA was first described by Theodor Leber in the 19th century. [7] [8] It should not be confused with Leber's hereditary optic neuropathy, which is a different disease also described by Theodor Leber.

Theodor Karl Gustav von Leber was a German ophthalmologist from Karlsruhe.
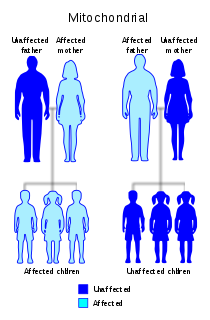
Leber's hereditary optic neuropathy (LHON) is a mitochondrially inherited degeneration of retinal ganglion cells (RGCs) and their axons that leads to an acute or subacute loss of central vision; this affects predominantly young adult males. LHON is only transmitted through the mother, as it is primarily due to mutations in the mitochondrial genome, and only the egg contributes mitochondria to the embryo. LHON is usually due to one of three pathogenic mitochondrial DNA (mtDNA) point mutations. These mutations are at nucleotide positions 11778 G to A, 3460 G to A and 14484 T to C, respectively in the ND4, ND1 and ND6 subunit genes of complex I of the oxidative phosphorylation chain in mitochondria. Men cannot pass on the disease to their offspring.















