In organic chemistry, a cycloalkyne is the cyclic analog of an alkyne (−C≡C−). A cycloalkyne consists of a closed ring of carbon atoms containing one or more triple bonds. Cycloalkynes have a general formula CnH2n−4. Because of the linear nature of the C−C≡C−C alkyne unit, cycloalkynes can be highly strained and can only exist when the number of carbon atoms in the ring is great enough to provide the flexibility necessary to accommodate this geometry. Large alkyne-containing carbocycles may be virtually unstrained, while the smallest constituents of this class of molecules may experience so much strain that they have yet to be observed experimentally. [1] Cyclooctyne (C8H12) is the smallest cycloalkyne capable of being isolated and stored as a stable compound. [2] Despite this, smaller cycloalkynes can be produced and trapped through reactions with other organic molecules or through complexation to transition metals.

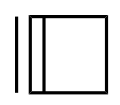
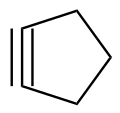


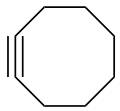



Due to the significant geometric constraints imposed by the R−C≡C−R functionality, cycloalkynes smaller than cyclodecyne (C10H16) result in highly strained structures. Cyclononyne (C9H14) and cyclooctyne (C8H12) are isolable (though strongly reactive) compounds. Cycloheptyne (C7H10), cyclohexyne (C6H8) and cyclopentyne (C5H6) only exist as transient reaction intermediates or ligands coordinating to a metal center. [3] Only a single osmium complex supports the potential existence of cyclobutyne (C4H4). [4] Cyclopropyne (C3H2) is entirely unknown.
Initial studies which demonstrated the transient intermediacy of the seven-, six- and five-membered cycloalkynes relied on trapping of the high-energy alkyne with a suitable reaction partner, such as a cyclic dienes or diazo compounds to generate the Diels–Alder or diazoalkane 1,3-dipolar cycloaddition products, respectively. [5] Stable small-ring cycloalkynes have subsequently been isolated in complex with various transition metals such as nickel, palladium and platinum. [6] Despite long being considered to be chemical curiosities with limited synthetic applications, recent work has demonstrated the utility of strained cycloalkynes in both total synthesis of complex natural products and bioorthogonal chemistry. [7] [8]
Angle strain in cycloalkynes arises from the deformation of the R−C≡C bond angle which must occur in order to accommodate the molecular geometry of rings containing less than ten carbons. The strain energies associated with cyclononyne (C9H14) and cyclooctyne (C8H12) are approximately 2.9 kcal/mol and 10 kcal/mol, respectively. [9] This upwards trend in energy for the isolable constituents of this class is indicative of a rapid escalation of angle strain with an inverse correlation to ring size. Analysis by photoelectron spectroscopy has indicated that the alkyne bond in small cyclic systems is composed of two non-degenerate π bonds – a highly reactive strained bond perpendicular to a lower-energy π bond. [10] Cis-bending of the R−C≡C bond angle results in the drastic lowering of the energy of the lowest unoccupied molecular orbital, a phenomenon which accounts for the reactivity of strained cycloalkynes from the perspective of molecular orbital theory. [11]
Initial efforts toward the synthesis of strained cycloalkynes showed that cycloalkynes could be generated via the elimination of hydrochloric acid from 1-chloro-cycloalkene in modest yield. The desired product could be obtained as a mixture with the corresponding allene as the major product. [12]
Further work in this area was aimed at developing milder reaction conditions and generating more robust yields. To circumvent the generation of the undesired allene, the Kobayashi method for aryne generation was adapted for the synthesis of cycloalkynes. [13]
More recently, a superior method for generating strained cycloalkynes was developed by Fujita. It involves base induced β-elimination of vinyl iodonium salts. The vinyl iodonium proved to be a particularly useful synthetic precursor to strained cycloalkynes due to its high reactivity which arises from the strong electron withdrawing ability of the positively charged iodine species as well as the leaving group ability of the iodonium. [14]
In addition to the elimination-type pathways described, cycloalkynes can also be obtained through the oxidation of cyclic bishydrazones with mercury oxide [15] or lead tetraacetate [3] as well as through the thermal decomposition of selenadiazole. [16]
Strained cycloalkynes are able to undergo all addition reactions typical to open chain alkynes. Due to the activated nature of the cyclic carbon–carbon triple bond, many alkyne addition-type reactions such as the Diels–Alder, 1,3-dipolar cycloadditions and halogenation may be performed using very mild conditions and in the absence of the catalysts frequently required to accelerate the transformation in a non-cyclic system. In addition to alkyne reactivity, cycloalkynes are able to undergo a number of unique, synthetically useful transformations.
One particularly intriguing mode of reactivity is the ring insertion of cyclohexyne into cyclic ketones. The reaction is initiated by the alkoxide-mediated generation of the reactive cycloalkyne species in situ, followed by the α-deprotonation of the ketone to yield the corresponding enolate. The two compounds then undergo a formal [2+2]-photocycloaddition to yield a highly unstable cyclobutanolate intermediate which readily decomposes to the enone product. [17]
This reaction was utilized as the key step in Carreira's total synthesis of guanacastapenes O and N. It allowed for the expedient construction of the 5-7-6 ring system and provided useful synthetic handles for subsequent functionalization. [18] [19]
Cyclooctyne, the smallest isolable cycloalkyne, is able to undergo azide-alkyne Huisgen cycloaddition under mild, physiological conditions in the absence of a copper(I) catalyst due to strain. This reaction has found widespread application as a bioorthogonal transformation for live cell imaging. [20] Although the mild, copper-catalyzed variant of the reaction, CuAAC (copper-catalyzed azide–alkyne cycloaddition) with linear alkynes had been known, development of the copper-free reaction was significant in that it provided facile reactivity while eliminating the need for a toxic metal catalyst. [21]
| Authority control databases: National |
|---|