| Ataxia-telangiectasia | |
|---|---|
| Specialty | Neurology, Medical genetics |
Ataxia telangiectasia (AT or A-T), also referred to as ataxia telangiectasia syndrome or Louis–Bar syndrome, [1] is a rare, neurodegenerative, autosomal recessive disease causing severe disability. Ataxia refers to poor coordination and telangiectasia to small dilated blood vessels, both of which are hallmarks of the disease. [2]
Ataxia is a neurological sign consisting of lack of voluntary coordination of muscle movements that can include gait abnormality, speech changes, and abnormalities in eye movements. Ataxia is a clinical manifestation indicating dysfunction of the parts of the nervous system that coordinate movement, such as the cerebellum. Ataxia can be limited to one side of the body, which is referred to as hemiataxia. Several possible causes exist for these patterns of neurological dysfunction. Dystaxia is a mild degree of ataxia. Friedreich's ataxia has gait abnormality as the most commonly presented symptom. The word is from Greek α- [a negative prefix] + -τάξις [order] = "lack of order".
![Telangiectasia small dilated blood vessels[1] near the surface of the skin or mucous membranes](https://upload.wikimedia.org/wikipedia/commons/thumb/2/2d/Case_115.jpg/320px-Case_115.jpg)
Telangiectasias, also known as spider veins, are small dilated blood vessels near the surface of the skin or mucous membranes, measuring between 0.5 and 1 millimeter in diameter.
Contents
- Symptoms
- Ataxia and other neurologic problems
- Telangiectasia
- Immune problems
- Cancer
- Skin
- Lung disease
- Feeding, swallowing, and nutrition
- Eye and vision
- Orthopedics
- Pathophysiology
- Cancer and radiosensitivity
- Delayed pubertal development (gonadal dysgenesis)
- Immune system defects and immune-related cancers
- Progeric changes
- Telangiectasia 2
- Increased alpha-fetoprotein (AFP) levels
- Neurodegeneration
- Radiation exposure
- Genetics and information about A-T carriers
- Diagnosis
- Differential diagnosis
- Management
- Ataxia and other neurologic problems 2
- Immune problems 2
- Lung disease 2
- Feeding, swallowing and nutrition
- Education and socialization
- Clinics and support
- Epidemiology
- Prognosis
- Research directions
- References
- External links
A-T affects many parts of the body:
- It impairs certain areas of the brain including the cerebellum, causing difficulty with movement and coordination.
- It weakens the immune system, causing a predisposition to infection.
- It prevents repair of broken DNA, increasing the risk of cancer.

The cerebellum is a major feature of the hindbrain of all vertebrates. Although usually smaller than the cerebrum, in some animals such as the mormyrid fishes it may be as large as or even larger. In humans, the cerebellum plays an important role in motor control. It may also be involved in some cognitive functions such as attention and language as well as in regulating fear and pleasure responses, but its movement-related functions are the most solidly established. The human cerebellum does not initiate movement, but contributes to coordination, precision, and accurate timing: it receives input from sensory systems of the spinal cord and from other parts of the brain, and integrates these inputs to fine-tune motor activity. Cerebellar damage produces disorders in fine movement, equilibrium, posture, and motor learning in humans.

The immune system is a host defense system comprising many biological structures and processes within an organism that protects against disease. To function properly, an immune system must detect a wide variety of agents, known as pathogens, from viruses to parasitic worms, and distinguish them from the organism's own healthy tissue. In many species, the immune system can be classified into subsystems, such as the innate immune system versus the adaptive immune system, or humoral immunity versus cell-mediated immunity. In humans, the blood–brain barrier, blood–cerebrospinal fluid barrier, and similar fluid–brain barriers separate the peripheral immune system from the neuroimmune system, which protects the brain.
Symptoms most often first appear in early childhood (the toddler stage) when children begin to sit or walk. Though they usually start walking at a normal age, they wobble or sway when walking, standing still or sitting. In late pre-school and early school age, they develop difficulty moving their eyes in a natural manner from one place to the next (oculomotor apraxia). They develop slurred or distorted speech, and swallowing problems. Some have an increased number of respiratory tract infections (ear infections, sinusitis, bronchitis, and pneumonia). Because not all children develop in the same manner or at the same rate, it may be some years before A-T is properly diagnosed. Most children with A-T have stable neurologic symptoms for the first 4–5 years of life, but begin to show increasing problems in early school years.
Oculomotor apraxia (OMA), also known as Cogan ocular motor apraxia or saccadic initiation failure (SIF) is the absence or defect of controlled, voluntary, and purposeful eye movement. It was first described in 1952 by the American ophthalmologist David Glendenning Cogan. People with this condition have difficulty moving their eyes horizontally and moving them quickly. The main difficulty is in saccade initiation, but there is also impaired cancellation of the vestibulo-ocular reflex. Patients have to turn their head in order to compensate for the lack of eye movement initiation in order to follow an object or see objects in their peripheral vision, but they often exceed their target. There is controversy regarding whether OMA should be considered an apraxia, since apraxia is the inability to perform a learned or skilled motor action to command, and saccade initiation is neither a learned nor a skilled action.

Sinusitis, also known as a sinus infection or rhinosinusitis, is inflammation of the mucous membrane that lines the sinuses resulting in symptoms. Common symptoms include thick nasal mucus, a plugged nose, and facial pain. Other signs and symptoms may include fever, headaches, poor sense of smell, sore throat, and cough. The cough is often worse at night. Serious complications are rare. It is defined as acute sinusitis if it lasts less than 4 weeks, and as chronic sinusitis if it lasts for more than 12 weeks.
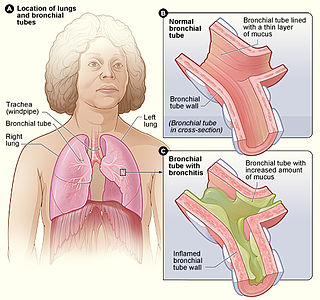
Bronchitis is inflammation of the bronchi in the lungs. Symptoms include coughing up mucus, wheezing, shortness of breath, and chest discomfort. Bronchitis is divided into two types: acute and chronic. Acute bronchitis is also known as a chest cold.
A-T is caused by a defect in the ATM gene, [3] The protein produced by the ATM gene recognizes that there is a break in dsDNA, recruits other proteins to fix the break, and stops the cell from making new DNA until the repair is complete. [4] The prevalence of A-T is estimated to be as high as 1 in 40,000 to as low as 1 in 300,000 people. [5] [6]

ATM serine/threonine kinase, symbol ATM, is a serine/threonine protein kinase that is recruited and activated by DNA double-strand breaks. It phosphorylates several key proteins that initiate activation of the DNA damage checkpoint, leading to cell cycle arrest, DNA repair or apoptosis. Several of these targets, including p53, CHK2, BRCA1, NBS1 and H2AX are tumor suppressors.











