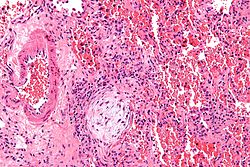
Bronchiolitis obliterans with organizing pneumonia, idiopathic interstitial pneumonia[1]
Micrograph showing a Masson body (off center left/bottom of the image – pale circular and paucicellular), as may be seen in cryptogenic organizing pneumonia. The Masson body plugs the airway. The artery associated with the obliterated airway is also seen (far left of the image). H&E stain.
It is often a complication of an existing chronic inflammatory disease such as rheumatoid arthritis, dermatomyositis, or it can be a side effect of certain medications such as amiodarone. COP was first described by Gary Epler in 1985.[5]
"Organizing" refers to unresolved pneumonia (in which the alveolar exudate persists and eventually undergoes fibrosis) in which fibrous tissue forms in the alveoli. The phase of resolution and/or remodeling following bacterial infections is commonly referred to as organizing pneumonia, both clinically and pathologically.
The American Thoracic Society and the European Respiratory Society hold that "cryptogenic organizing pneumonia" is the preferred clinical term for this disease for multiple reasons:[6][7]
Avoid confusion with bronchiolitis obliterans, which may not be visualized in every case of this disease.
The classic presentation of COP is the development of nonspecific systemic (e.g., fevers, chills, night sweats, fatigue, weight loss) and respiratory (e.g. difficulty breathing, cough) symptoms in association with filling of the lung alveoli that is visible on chest x-ray.[8] This presentation is usually so suggestive of an infection that the majority of patients with COP have been treated with at least one failed course of antibiotics by the time the true diagnosis is made.[8] Symptoms are usually subacute, occurring over weeks to months with dry cough (seen in 71% of people), dyspnea (shortness of breath)(62%) and fever (44%) being the most common symptoms.[9]
Vaping: On October 17, 2019, the American Journal of Clinical Pathology reported that lung biopsies from patients with vaping-associated pulmonary illness show acute lung injury patterns, including organizing pneumonia.[11]
Analysis of COVID-19 CT imaging along with postmortem lung biopsies and autopsies suggest that the majority of patients with COVID-19 pulmonary involvement also have secondary organizing pneumonia (OP) or its histological variant, acute fibrinous and organizing pneumonia, which are both well-known complications of viral infections.[15]
It was identified in 1985, although its symptoms had been noted before but not recognised as a separate lung disease. The risk of COP is higher for people with inflammatory diseases like lupus, dermatomyositis, rheumatoid arthritis, and scleroderma.[16] It most commonly presents in the 5th or 6th decade of life and it is exceedingly rare in children.[9][17]
Pathophysiology
Organizing pneumonia is usually preceded by some type of lung injury that causes a localized denudation or disruption in continuity of the epithelial basal laminae of the type 1 alveolar pneumocytes that line the alveoli.[9] This injury to the epithelial basal lamina results in inflammatory cells and plasma proteins leaking into the alveolar space and forming fibrin, resulting in an initial fibroblast driven intra-alveolar fibroproliferation.[9] The fibroblasts differentiate into myofibroblasts and continue to form fibrosis resulting in intra-alveolar fibroinflammatory buds (Masson's Bodies) that are characteristic of organizing pneumonia.[9] These Masson's bodies consist of inflammatory cells contained in an extracellular matrix consisting of type I collagen, fibronectin, procollagen type III, tenascin C and proteoglycans.[9]Angiogenesis , or the formation of blood vessels, occurs in the Masson's bodies and this is driven by vascular endothelial growth factor.[9] Remodeling occurs, resulting in the intra-alveolar fibroinflammatory buds (Masson's Bodies) moving into the interstitial space and forming collagen globules that are then covered by type 1 alveolar epithelial cells with well developed basement membranes. These type 1 alveolar epithelial cells (pneumocytes) then proliferate, restoring the continuity and function of the alveolar unit.[9] This process is in contrast to the histopathologic changes seen in usual interstitial pneumonia where extensive fibrosis and inflammation occur leading to fibroblastic foci to form in the alveolar spaces resulting in obliteration of the alveolar space, scarring and significant damage to lung architecture (the alveoli).[9]
Tissue inhibitors of metalloproteinases (which inhibit breakdown of the extracellular matrix connective tissue) are more active in usual interstitial pneumonia as compared to organizing pneumonia, this is thought to lead to a greater deposition of connective tissue in the alveolar space in interstitial pneumonia as compared to organizing pneumonia and may explain the progressive, irreversible fibrosis seen in usual interstitial pneumonia.[9] Gelatinolytic activity (resulting in the breakdown of extracellular matrix connective tissue) is greater in organizing pneumonia as compared to usual interstitial pneumonia, and this is thought to contribute to the reversible fibroproliferation characteristic of organizing pneumonia.[9]
Pulmonary function testing in people with organizing pneumonia, either cryptogenic or due to secondary causes, shows a restrictive defect with a decrease in the gas absorptive capacity of the lungs (seen as a decrease in the diffusion capacity of carbon monoxide).[9] Airflow obstruction is usually not seen on pulmonary function testing.[9]
Bronchoscopy with bronchoalveolar lavage is recommended in possible cases of organizing pneumonia to rule out infection and other causes of alveolar infiltrates.[9] The bronchoalveolar lavage in organizing pneumonia shows a lymphocytic predominant inflammation of the alveoli with increases in neutrophils and eosinophils.[9] Resolution of inflammatory cells in the bronchoalveolar lavage is usually delayed in organizing pneumonia, lagging behind clinical and radiographic improvement.[9]
Biopsy findings in patients with organizing pneumonia consist of loose connective tissue plugs involving the alveoli, alveolar ducts and bronchioles. The loose connective tissue plugs occupying the alveolar spaces often connect to other connective tissue plugs in nearby alveoli via the pores of Kohn creating a characteristic butterfly pattern on histology.[9] There is usually minimal to no interstitial inflammatory changes in biopsies of organizing pneumonia.[9]
Imaging
CT scan showing cryptogenic organizing pneumonia (biopsy-proven)The reversed halo sign is seen in about 20% of individuals with COP.
The chest x-ray is distinctive with features that appear similar to an extensive pneumonia, with both lungs showing widespread white patches. The white patches may seem to migrate from one area of the lung to another as the disease persists or progresses. Computed tomography (CT) may be used to confirm the diagnosis. Often the findings are typical enough to allow the doctor to make a diagnosis without ordering additional tests.[19] To confirm the diagnosis, a doctor may perform a lung biopsy using a bronchoscope. Many times, a larger specimen is needed and must be removed surgically.
Plain chest radiography shows normal lung volumes, with characteristic patchy unilateral or bilateral consolidation. Small nodular opacities occur in up to 50% of patients and large nodules in 15%. On high resolution computed tomography, airspace consolidation with air bronchograms is present in more than 90% of patients, often with a lower zone predominance. A subpleural or peribronchiolar distribution is noted in up to 50% of patients. Ground glass appearance or hazy opacities associated with the consolidation are detected in most patients.
Histologically, cryptogenic organizing pneumonia is characterized by the presence of polypoid plugs of loose organizing connective tissue (Masson bodies) within alveolar ducts, alveoli, and bronchioles.
Unusual presentations of organizing pneumonia
While patchy bilateral disease is typical, there are unusual variants of organizing pneumonia where it may appear as multiple nodules or masses. One rare presentation, focal organizing pneumonia, may be indistinguishable from lung cancer based on imaging alone, requiring biopsy or surgical resection to make the diagnosis.[20]
Complications
Rare cases of COP have induced with lobar cicatricial atelectasis.[21]
Treatment
Systemic steroids are considered the first line treatment for organizing pneumonia, with patient's often having clinical improvement within 72 hours of steroid initiation and most patients achieving recovery.[22][9] A prolonged treatment course is indicated, with patients usually requiring at least 4–6 months of treatment.[9] Patients who are treated with larger doses of steroids require prophylaxis against pneumocystis jirovecii.[9] Relapses may occur and are more likely to occur in severe disease or when steroids are tapered too soon or too quickly.[9] Alternative or adjunct treatment options include macrolide antibiotics (due to anti-inflammatory properties), azathioprine and cyclophosphamide.[9]
↑ Epler GR (June 2011). "Bronchiolitis obliterans organizing pneumonia, 25 years: a variety of causes, but what are the treatment options?". Expert Rev Respir Med. 5 (3): 353–61. doi:10.1586/ers.11.19. PMID21702658. S2CID207222916.
↑ Joseph F. Tomashefski; Carol Farver; Armando E. Fraire (2009). Dail and Hammar's Pulmonary Pathology: Volume I: Nonneoplastic Lung Disease (3rded.). Springer Science & Business Media. p.64. ISBN978-0-387-68792-6.
↑ Mukhopadhyay, Sanjay; Mehrad, Mitra; Dammert, Pedro; Arrossi, Andrea V; Sarda, Rakesh; Brenner, David S; Maldonado, Fabien; Choi, Humberto; Ghobrial, Michael (2019). "Lung Biopsy Findings in Severe Pulmonary Illness Associated With E-Cigarette Use (Vaping): A Report of Eight Cases". American Journal of Clinical Pathology. 153 (1): 30–39. doi:10.1093/ajcp/aqz182. ISSN0002-9173. PMID31621873.
↑ Radzikowska, E; Nowicka, U; Wiatr, E; Jakubowska, L; Langfort, R; Chabowski, M; Roszkowski, K (2007). "Organising pneumonia and lung cancer - case report and review of the literature". Pneumonologia I Alergologia Polska. 75 (4): 394–7. PMID18080991.
↑ Yoshida, K; Nakajima, M; Niki, Y; Matsushima, T (2001). "Atelectasis of the right lower lobe in association with bronchiolitis obliterans organizing pneumonia". Nihon Kokyuki Gakkai Zasshi = The Journal of the Japanese Respiratory Society. 39 (4): 260–5. PMID11481825.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.