Kawasaki disease (also known as mucocutaneous lymph node syndrome) is a syndrome of unknown cause that results in a fever and mainly affects children under 5 years of age.[6] It is a form of vasculitis, in which medium-sized blood vessels become inflamed throughout the body.[1] The fever typically lasts for more than five days and is not affected by usual medications.[1] Other common symptoms include large lymph nodes in the neck, a rash in the genital area, lips, palms, or soles of the feet, and red eyes.[1] Within three weeks of the onset, the skin from the hands and feet may peel, after which recovery typically occurs.[1] The disease is the leading cause of acquired heart disease in children in developed countries, which include the formation of coronary artery aneurysms and myocarditis.[1][7]
While the specific cause is unknown, it is thought to result from an excessive immune response to particular infections in children who are genetically predisposed to those infections.[6] It is not an infectious disease, that is, it does not spread between people.[8] Diagnosis is usually based on a person's signs and symptoms.[1] Other tests such as an ultrasound of the heart and blood tests may support the diagnosis.[1] Diagnosis must take into account many other conditions that may present similar features, including scarlet fever and juvenile rheumatoid arthritis.[9]Multisystem inflammatory syndrome in children, a "Kawasaki-like" disease associated with COVID-19,[10] appears to have distinct features.[11][12] Another disease that can originally be presented with the same symptoms is measles. This is due to similar symptoms such as red eyes, persistent fever, and swollen hands/feet. However, Kawasaki disease is not spread the way measles can be spread from person to person.[13]
Typically, initial treatment of Kawasaki disease consists of high doses of aspirin and immunoglobulin.[1] Usually, with treatment, fever resolves within 24 hours and full recovery occurs.[1] If the coronary arteries are involved, ongoing treatment or surgery may occasionally be required.[1] Without treatment, coronary artery aneurysms occur in up to 25% and about 1% die.[4][14] With treatment, the risk of death is reduced to 0.17%.[14] People who have had coronary artery aneurysms after Kawasaki disease require lifelong cardiological monitoring by specialized teams.[15]
Kawasaki disease is rare.[1] It affects between 8 and 67 per 100,000 people under the age of five except in Japan, where it affects 124 per 100,000.[5] Boys are more commonly affected than girls.[1] The disorder is named after Japanese pediatricianTomisaku Kawasaki, who first described it in 1967.[5][16]
Signs and symptoms
Signs of Kawasaki disease
Kawasaki disease often begins with a high and persistent fever that is not very responsive to normal treatment with paracetamol (acetaminophen) or ibuprofen.[17][18] This is the most prominent symptom of Kawasaki disease, and is a characteristic sign that the disease is in its acute phase; the fever normally presents as a high (above 39–40°C) and remittent, and is followed by extreme irritability.[18][19] Recently, it is reported to be present in patients with atypical or incomplete Kawasaki disease;[20][21] nevertheless, it is not present in 100% of cases.[22]
The first day of fever is considered the first day of the illness,[17] and its duration is typically one to two weeks; in the absence of treatment, it may extend for three to four weeks.[4] Prolonged fever is associated with a higher incidence of cardiac involvement.[23] It responds partially to antipyretic drugs and does not cease with the introduction of antibiotics.[4] However, when appropriate therapy is started– intravenous immunoglobulin and aspirin– the fever subsides after two days.[24]
Bilateral conjunctival inflammation has been reported to be the most common symptom after fever.[25][26] It typically involves the bulbar conjunctivae, is not accompanied by suppuration, and is not painful.[27] This usually begins shortly after the onset of fever during the acute stage of the disease.[17]Anterior uveitis may be present under slit-lamp examination.[28][29]Iritis can occur, too.[30]Keratic precipitates are another eye manifestation (detectable by a slit lamp, but are usually too small to be seen by the unaided eye).[17][31]
Cervical lymphadenopathy is seen in 50% to 75% of children, whereas the other features are estimated to occur in 90%,[17][25] but sometimes it can be the dominant presenting symptom.[31][34] According to the diagnostic criteria, at least one impaired lymph node ≥ 15mm in diameter should be involved.[33] Affected lymph nodes are painless or minimally painful, nonfluctuant, and nonsuppurative; erythema of the neighboring skin may occur.[17] Children with fever and neck adenitis who do not respond to antibiotics should have Kawasaki disease considered as part of the differential diagnoses.[17]
In the acute phase of the disease, changes in the peripheral extremities can include erythema of the palms and soles, which is often striking with sharp demarcation[17] and often accompanied by painful, brawny edema of the dorsa of the hands or feet, so affected children frequently refuse to hold objects in their hands or to bear weight on their feet.[4][17] Later, during the convalescent or the subacute phase, desquamation of the fingers and toes usually begins in the periungual region within two to three weeks after the onset of fever and may extend to include the palms and soles.[38] Around 11% of children affected by the disease may continue skin-peeling for many years.[39] One to two months after the onset of fever, deep transverse grooves across the nails may develop (Beau's lines),[40] and occasionally nails are shed.[40]
The most common skin manifestation is a diffuse macular-papular erythematous rash, which is quite nonspecific.[41] The rash varies over time and is characteristically located on the trunk; it may further spread to involve the face, extremities, and perineum.[4] Many other forms of cutaneous lesions have been reported; they may include scarlatiniform, papular, urticariform, multiform-like erythema, and purpuric lesions; even micropustules were reported.[42][43] It can be polymorphic, not itchy, and normally observed up to the fifth day of fever.[44] However, it is never bullous or vesicular.[4]
In the acute stage of Kawasaki disease, systemic inflammatory changes are evident in many organs.[45] Joint pain (arthralgia) and swelling, frequently symmetrical, and arthritis can also occur.[25]Myocarditis,[46]diarrhea,[33]pericarditis, valvulitis, aseptic meningitis, pneumonitis, lymphadenitis, and hepatitis may be present and are manifested by the presence of inflammatory cells in the affected tissues.[45] If left untreated, some symptoms will eventually relent, but coronary artery aneurysms will not improve, resulting in a significant risk of death or disability due to myocardial infarction.[33] If treated quickly, this risk can be mostly avoided and the course of illness cut short.[47]
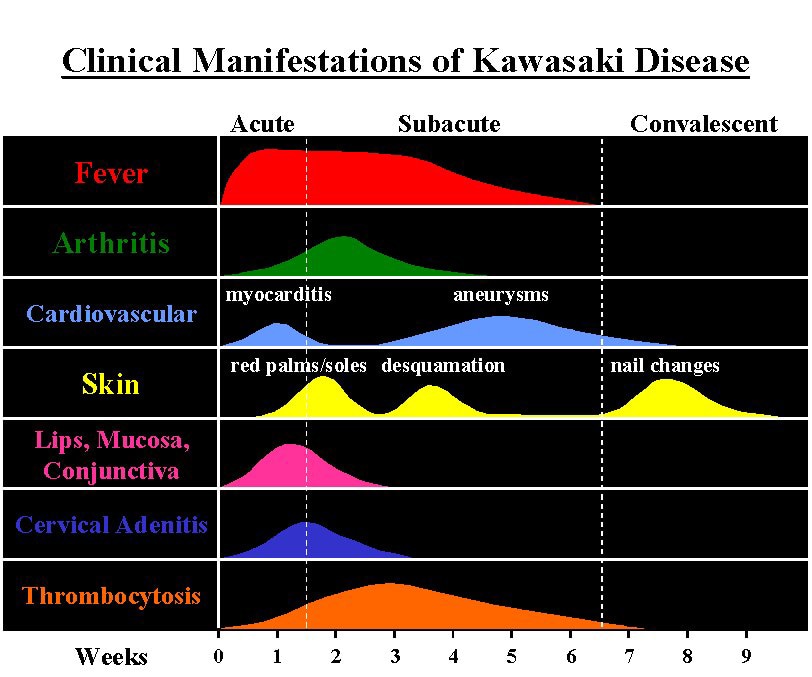
Signs and symptoms and time course of Kawasaki disease
The course of the disease can be divided into three clinical phases.[49]
The acute febrile phase, which usually lasts for one to two weeks, is characterized by fever, conjunctival injection, erythema of the oral mucosa, erythema and swelling of the hands and feet, rash, cervical adenopathy, aseptic meningitis, diarrhea, and hepatic dysfunction.[33] Myocarditis is common during this time, and a pericardial effusion may be present.[17]Coronary arteritis may be present, but aneurysms are generally not yet visible by echocardiography.
The subacute phase begins when fever, rash, and lymphadenopathy resolve at about one to two weeks after the onset of fever, but irritability, anorexia, and conjunctival injection persist. Desquamation of the fingers and toes and thrombocytosis are seen during this stage, which generally lasts until about four weeks after the onset of fever. Coronary artery aneurysms usually develop during this time, and the risk for sudden death is highest.[17][50]
The convalescent stage begins when all clinical signs of illness have disappeared, and continues until the sedimentation rate returns to normal, usually at six to eight weeks after the onset of illness.[33]
Adult onset of Kawasaki disease is rare.[51] The presentation differs between adults and children: in particular, it seems that adults more often have cervical lymphadenopathy, hepatitis, and arthralgia.[33][51]
Some children, especially young infants,[52] have atypical presentations without the classic set of symptoms.[49] Such presentations are associated with a higher risk of cardiac artery aneurysms.[17][53]
Cardiac
X-ray showing aneurysmal enlargement of the coronary arteries, which is a complication in a Kawasaki syndrome
Heart complications are the most important aspect of Kawasaki disease, which is the leading cause of heart disease acquired in childhood in the United States and Japan.[33] In developed nations, it appears to have replaced acute rheumatic fever as the most common cause of acquired heart disease in children.[17] Coronary artery aneurysms occur as a sequela of the vasculitis in 20–25% of untreated children.[54] It is first detected at a mean of 10 days of illness and the peak frequency of coronary artery dilation or aneurysms occurs within four weeks of onset.[50] Aneurysms are classified into small (internal diameter of vessel wall <5mm), medium (diameter ranging from 5–8mm), and giant (diameter > 8mm).[33] Saccular and fusiform aneurysms usually develop between 18 and 25 days after the onset of illness.[17]
Even when treated with high-dose IVIG regimens within the first 10 days of illness, 5% of children with Kawasaki disease develop at the least transient coronary artery dilation and 1% develop giant aneurysms.[55][56][57] Death can occur either due to myocardial infarction secondary to blood clot formation in a coronary artery aneurysm or to rupture of a large coronary artery aneurysm. Death is most common two to 12 weeks after the onset of illness.[17]
Many risk factors predicting coronary artery aneurysms have been identified,[23] including persistent fever after IVIG therapy,[58][59] low hemoglobin concentrations, low albumin concentrations, high white-blood-cell count, high band count, high CRP concentrations, male sex, and age less than one year.[60] Coronary artery lesions resulting from Kawasaki disease change dynamically with time.[4] Resolution one to two years after the onset of the disease has been observed in half of vessels with coronary aneurysms.[61][62]Narrowing of the coronary artery, which occurs as a result of the healing process of the vessel wall, often leads to significant obstruction of the blood vessel and the heart not receiving enough blood and oxygen.[61] This can eventually lead to heart muscle tissue death, i.e., myocardial infarction (MI).[61]
MI caused by thrombotic occlusion in an aneurysmal, stenotic, or both aneurysmal and stenotic coronary artery is the main cause of death from Kawasaki disease.[63] The highest risk of MI occurs in the first year after the onset of the disease.[63] MI in children presents with different symptoms from those in adults. The main symptoms were shock, unrest, vomiting, and abdominal pain; chest pain was most common in older children.[63] Most of these children had the attack occurring during sleep or at rest, and around one-third of attacks were asymptomatic.[17]
Other Kawasaki disease complications have been described, such as aneurysm of other arteries: aortic aneurysm,[68] with a higher number of reported cases involving the abdominal aorta,[69][70]axillary artery aneurysm,[71]brachiocephalic artery aneurysm,[72] aneurysm of iliac and femoral arteries, and renal artery aneurysm.[4][73] Other vascular complications can occur such as increased wall thickness and decreased distensibility of carotid arteries,[74]aorta,[75] and brachioradial artery.[76] This change in the vascular tone is secondary to endothelial dysfunction.[73] In addition, children with Kawasaki disease, with or without coronary artery complications, may have a more adverse cardiovascular risk profile later in life,[76] and may benefit from long term monitoring and prevention approaches to both detect cardiovascular disease early such as thrombosis or stenosis and prevent onset.[15] Examples of prevention approaches include lipid lowering medications, blood pressure medication, smoking cessation, and healthy active living.[15]
Circumstantial evidence points to an infectious cause.[105] Since recurrences are unusual in Kawasaki disease, it is thought that the trigger is more likely to be represented by a single pathogen, rather than a range of viral or bacterial agents.[106] Various candidates have been implicated, including upper respiratory tract infection by some novel RNA virus.[6] Despite intensive search, no single pathogen has been identified.[104] There has been debate as to whether the infectious agent might be a superantigen (i.e. one commonly associated with excessive immune system activation).[102][107] Current consensus favors an excessive immunologic response to a conventional antigen which usually provides future protection.[6] Research points to an unidentified ubiquitous virus,[108] possibly one that enters through the respiratory tract.[109]
Seasonal trends in the appearance of new cases of Kawasaki disease have been linked to tropospheric wind patterns, which suggests wind-borne transport of something capable of triggering an immunologic cascade when inhaled by genetically susceptible children.[6] Winds blowing from central Asia correlate with numbers of new cases of Kawasaki disease in Japan, Hawaii, and San Diego.[110] These associations are themselves modulated by seasonal and interannual events in the El Niño–Southern Oscillation in winds and sea surface temperatures over the tropical eastern Pacific Ocean.[111] Efforts have been made to identify a possible pathogen in air-filters flown at altitude above Japan.[112] One source has been suggested in northeastern China.[6][113]
Genetics
Genetic susceptibility is suggested by increased incidence among children of Japanese descent around the world, and also among close and extended family members of affected people.[6] Genetic factors are also thought to influence development of coronary artery aneurysms and response to treatment.[114] The exact genetic contribution remains unknown.[115]Genome-wide association studies and studies of individual candidate genes have together helped identify specific single nucleotide polymorphisms (SNPs), mostly found in genes with immune regulatory functions.[114] The associated genes and their levels of expression appear to vary among different ethnic groups, both with Asian and non-Asian backgrounds.[116]
SNPs in FCGR2A, CASP3, BLK, ITPKC, CD40 and ORAI1 have all been linked to susceptibility, prognosis, and risk of developing coronary artery aneurysms.[116] Various other possible susceptibility genes have been proposed, including polymorphisms in the HLA region, but their significance is disputed.[115] Genetic susceptibility to Kawasaki disease appears complex.[117]Gene–gene interactions also seem to affect susceptibility and prognosis.[116] At an epigenetic level, altered DNA methylation has been proposed as an early mechanistic factor during the acute phase of the disease.[116]
Diagnosis
Criteria for diagnosis
Fever of ≥5 days' duration associated with at least four† of these five changes
Disease cannot be explained by some other known disease process
†A diagnosis of Kawasaki disease can be made if fever and only three changes are present if coronary artery disease is documented by two-dimensional echocardiography or coronary angiography.
Source: Nelson's essentials of pediatrics,[118] Review[119]
Since no specific laboratory test exists for Kawasaki disease, diagnosis must be based on clinical signs and symptoms, together with laboratory findings.[9] Timely diagnosis requires careful history-taking and thorough physical examination.[120] Establishing the diagnosis is difficult, especially early in the course of the illness, and frequently children are not diagnosed until they have seen several health-care providers. Many other serious illnesses can cause similar symptoms, and must be considered in the differential diagnosis, including scarlet fever, toxic shock syndrome, juvenile idiopathic arthritis, and childhood mercury poisoning (infantile acrodynia).[121]
Classically, five days of fever[122] plus four of five diagnostic criteria must be met to establish the diagnosis. The criteria are:[123]
erythema of the lips or oral cavity or cracking of the lips
rash on the trunk
swelling or erythema of the hands or feet
red eyes (conjunctival injection)
swollen lymph node in the neck of at least 15mm
Many children, especially infants, eventually diagnosed with Kawasaki disease, do not exhibit all of the above criteria. In fact, many experts now recommend treating for Kawasaki disease even if only three days of fever have passed and at least three diagnostic criteria are present, especially if other tests reveal abnormalities consistent with Kawasaki disease. In addition, the diagnosis can be made purely by the detection of coronary artery aneurysms in the proper clinical setting.[citation needed]
Investigations
A physical examination will demonstrate many of the features listed above.
Angiography was historically used to detect coronary artery aneurysms, and remains the gold standard for their detection, but is rarely used today unless coronary artery aneurysms have already been detected by echocardiography.
Biopsy is rarely performed, as it is not necessary for diagnosis.[8]
Subtypes
Based on clinical findings, a diagnostic distinction may be made between the "classic" or "typical" presentation of Kawasaki disease and "incomplete" or "atypical" presentation of a "suspected" form of the disease.[6] Regarding incomplete/atypical presentation, American Heart Association guidelines state that Kawasaki disease "should be considered in the differential diagnosis of prolonged unexplained fever in childhood associated with any of the principal clinical features of the disease, and the diagnosis can be considered confirmed when coronary artery aneurysms are identified in such patients by echocardiography."[6]
A further distinction between incomplete and atypical subtypes may also be made in the presence of non-typical symptoms.[49]
Case definition
For study purposes, including vaccine safety monitoring, an international case definition has been proposed to categorize 'definite' (i.e. complete/incomplete), 'probable' and 'possible' cases of Kawasaki disease.[125]
Several reported cases suggest that this Kawasaki-like multisystem inflammatory syndrome is not limited to children; there is the possibility of an analogous disease in adults, which has been termed MIS-A. Some suspected patients have presented with positive test results for SARS-CoV-2 and reports suggest intravenous immunoglobulin, anticoagulation, tocilizumab, plasmapheresis and steroids are potential treatments.[129][130][131]
Classification
Debate has occurred about whether Kawasaki disease should be viewed as a characteristic immune response to some infectious pathogen, as an autoimmune process, or as an autoinflammatory disease (i.e. involving innate rather than adaptive immune pathways).[102] Overall, immunological research suggests that Kawasaki disease is associated with a response to a conventional antigen (rather than a superantigen) that involves both activation of the innate immune system and also features of an adaptive immune response.[6][132] Identification of the exact nature of the immune process involved in Kawasaki disease could help guide research aimed at improving clinical management.[102]
Inflammation, or vasculitis, of the arteries and veins occurs throughout the body, usually caused by increased production of the cells of the immune system to a pathogen, or autoimmunity.[133] Systemic vasculitides may be classified according to the type of cells involved in the proliferation, as well as the specific type of tissue damage occurring within the vein or arterial walls.[133] Under this classification scheme for systemic vasculitis, Kawasaki disease is considered to be a necrotizing vasculitis (also called necrotizing angiitis), which may be identified histologically by the occurrence of necrosis (tissue death), fibrosis, and proliferation of cells associated with inflammation in the inner layer of the vascular wall.[133][134]
Kawasaki disease may be further classified as a medium-sized vessel vasculitis, affecting medium- and small-sized blood vessels,[45][135][136] such as the smaller cutaneous vasculature (veins and arteries in the skin) that range from 50 to 100μm in diameter.[33][137] Kawasaki disease is also considered to be a primary childhood vasculitis, a disorder associated with vasculitis that mainly affects children under the age of 18.[119][138] A recent, consensus-based evaluation of vasculitides occurring primarily in children resulted in a classification scheme for these disorders, to distinguish them and suggest a more concrete set of diagnostic criteria for each.[119] Within this classification of childhood vasculitides, Kawasaki disease is, again, a predominantly medium-sized vessel vasculitis.[119]
Children with Kawasaki disease should be hospitalized and cared for by a physician who has experience with this disease. In an academic medical center, care is often shared between pediatric cardiology, pediatric rheumatology, and pediatric infectious disease specialists (although no specific infectious agent has yet been identified).[141] To prevent damage to coronary arteries, treatment should be started immediately following the diagnosis.[citation needed]
Intravenous immunoglobulin (IVIG) is the standard treatment for Kawasaki disease[142] and is administered in high doses with marked improvement usually noted within 24 hours. If the fever does not respond, an additional dose may be considered. In rare cases, a third dose may be given. IVIG is most useful within the first seven days of fever onset, to prevent coronary artery aneurysm. IVIG given within the first 10 days of the disease reduces the risk of damage to the coronary arteries in children, without serious adverse effects.[142] A 2023 systematic review and meta-analysis revealed that no prediction models of IVIG resistance in patients with Kawasaki disease could accurately distinguish the resistance.[143] The largest clinical study to date on treatment of resistant Kawasaki disease has shown that infliximab is more effective and safer than a second dose of IVIG in treating children with IVIG-resistant Kawasaki Disease, offering faster fever resolution and reduced hospitalization times without significant adverse events.[144]
Salicylate therapy, particularly aspirin, remains an important part of the treatment (though questioned by some)[145] but salicylates alone are not as effective as IVIG. There is limited evidence to indicate whether children should continue to receive salicylate as part of their treatment.[146] Aspirin therapy is started at high doses until the fever subsides, and then is continued at a low dose when the patient returns home, usually for two months to prevent blood clots from forming. Except for Kawasaki disease and a few other indications, aspirin is otherwise normally not recommended for children due to its association with Reye syndrome. Because children with Kawasaki disease will be taking aspirin for up to several months, vaccination against varicella and influenza is required, as these infections are most likely to cause Reye syndrome.[147]
High-dose aspirin is associated with anemia and does not confer benefit to disease outcomes.[148]
About 15-20% of children following the initial IVIG infusion show persistent or recurrent fever and are classified as IVIG-resistant. While the use of TNF alpha blockers (TNF-α) may reduce treatment resistance and the infusion reaction after treatment initiation, further research is needed.[149] Due to the potential involvement of the upregulated calcium-nuclear factor of activated T cells pathway in the development of the disease, a 2019 study found that the combination of ciclosporin and IVIG infusion can suppress coronary artery abnormalities. Further research is needed to determine which patients would respond best to this treatment.[150]
Corticosteroids have also been used,[151] especially when other treatments fail or symptoms recur, but in a randomized controlled trial, the addition of corticosteroid to immune globulin and aspirin did not improve outcome.[152] Additionally, corticosteroid use in the setting of Kawasaki disease is associated with increased risk of coronary artery aneurysm, so its use is generally contraindicated in this setting. In cases of Kawasaki disease refractory to IVIG, cyclophosphamide and plasma exchange have been investigated as possible treatments, with variable outcomes. However, a Cochrane review published in 2017 (updated in 2022) found that, in children, the use of corticosteroids in the acute phase of KD was associated with improved coronary artery abnormalities, shorter hospital stays, a decreased duration of clinical symptoms, and reduced inflammatory marker levels. Patient populations based in Asia, people with higher risk scores, and those receiving longer steroid treatment may have greater benefit from steroid use.[153]
Prognosis
With early treatment, rapid recovery from the acute symptoms can be expected, and the risk of coronary artery aneurysms is greatly reduced. Untreated, the acute symptoms of Kawasaki disease are self-limited (i.e. the patient will recover eventually), but the risk of coronary artery involvement is much greater, even many years later. Many cases of myocardial infarction in young adults have now been attributed to Kawasaki disease that went undiagnosed during childhood.[6] Overall, about 2% of patients die from complications of coronary vasculitis.[citation needed]
Laboratory evidence of increased inflammation combined with demographic features (male sex, age less than six months or greater than eight years) and incomplete response to IVIG therapy create a profile of a high-risk patient with Kawasaki disease.[60][154] The likelihood that an aneurysm will resolve appears to be determined in large measure by its initial size, in which the smaller aneurysms have a greater likelihood of regression.[155][156] Other factors are positively associated with the regression of aneurysms, including being younger than a year old at the onset of Kawasaki disease, fusiform rather than saccular aneurysm morphology, and an aneurysm location in a distal coronary segment.[62] The highest rate of progression to stenosis occurs among those who develop large aneurysms.[4] The worst prognosis occurs in children with giant aneurysms.[157] This severe outcome may require further treatment such as percutaneous transluminal angioplasty,[158]coronary artery stenting,[159]bypass grafting,[160] and even cardiac transplantation.[161]
A relapse of symptoms may occur soon after initial treatment with IVIG. This usually requires rehospitalization and retreatment. Treatment with IVIG can cause allergic and nonallergic acute reactions, aseptic meningitis, fluid overload, and rarely, other serious reactions. Overall, life-threatening complications resulting from therapy for Kawasaki disease are exceedingly rare, especially compared with the risk of nontreatment. Evidence indicates Kawasaki disease produces altered lipid metabolism that persists beyond the clinical resolution of the disease.[citation needed]
Rarely, recurrence can occur in Kawasaki disease with or without treatment.[162][163]
Epidemiology
Kawasaki disease affects boys more than girls, with people of Asian ethnicity, particularly Japanese people. The higher incidence in Asian populations is thought to be linked to genetic susceptibility.[164] Incidence rates vary between countries.
Currently, Kawasaki disease is the most commonly diagnosed pediatric vasculitis in the world. By far, the highest incidence of Kawasaki disease occurs in Japan, with the most recent study placing the attack rate at 218.6 per 100,000 children less than five years of age (about one in 450 children). At this present attack rate, more than one in 150 children in Japan will develop Kawasaki disease during their lifetimes.[citation needed]
However, its incidence in the United States is increasing. Kawasaki disease is predominantly a disease of young children, with 80% of patients younger than five years of age. About 2,000–4,000 cases are identified in the U.S. each year (9 to 19 per 100,000 children younger than five years of age).[141][165][166] In the continental United States, Kawasaki disease is more common during the winter and early spring, boys with the disease outnumber girls by ≈1.5–1.7:1, and 76% of affected children are less than 5 years of age.[167]
In the United Kingdom, before 2000, it was diagnosed in fewer than one in every 25,000 people per year.[168] Incidence of the disease doubled from 1991 to 2000, however, with four cases per 100,000 children in 1991 compared with a rise of eight cases per 100,000 in 2000.[169] By 2017, this figure had risen to 12 in 100,000 people with 419 diagnosed cases of Kawasaki disease in the United Kingdom.[170]
In Japan, the rate is 240 in every 100,000 people.[171]
Coronary artery aneurysms due to Kawasaki disease are believed to account for 5% of acute coronary syndrome cases in adults under 40 years of age.[6]
History
The disease was first reported by Tomisaku Kawasaki in a four-year-old child with a rash and fever at the Red Cross Hospital in Tokyo in January 1961, and he later published a report on 50 similar cases.[16] Later, Kawasaki and colleagues were persuaded of definite cardiac involvement when they studied and reported 23 cases, of which 11 (48%) patients had abnormalities detected by an electrocardiogram.[172] In 1974, the first description of this disorder was published in the English-language literature.[173] In 1976, Melish et al. described the same illness in 16 children in Hawaii.[174] Melish and Kawasaki had independently developed the same diagnostic criteria for the disorder, which are still used today to make the diagnosis of classic Kawasaki disease.
A question was raised whether the disease only started during the period between 1960 and 1970, but later a preserved heart of a seven-year-old boy who died in 1870 was examined and showed three aneurysms of the coronary arteries with clots, as well as pathologic changes consistent with Kawasaki disease.[175] Kawasaki disease is now recognized worldwide. Why cases began to emerge across all continents around the 1960s and 1970s is unclear.[176] Possible explanations could include confusion with other diseases such as scarlet fever, and easier recognition stemming from modern healthcare factors such as the widespread use of antibiotics.[176] In particular, old pathological descriptions from Western countries of infantile polyarteritis nodosa coincide with reports of fatal cases of Kawasaki disease.[6]
In the United States and other developed nations, Kawasaki disease appears to have replaced acute rheumatic fever as the most common cause of acquired heart disease in children.[177]
12Kawasaki T (March 1967). "[Acute febrile mucocutaneous syndrome with lymphoid involvement with specific desquamation of the fingers and toes in children]". Arerugi. 16 (3): 178–222. PMID6062087.
↑Rowley AH, Gonzalez-Crussi F, Gidding SS, Duffy CE, Shulman ST (March 1987). "Incomplete Kawasaki disease with coronary artery involvement". The Journal of Pediatrics. 110 (3): 409–13. doi:10.1016/S0022-3476(87)80503-6. PMID3819942.
↑Rodriguez-Lozano AL, Rivas-Larrauri FE, Hernandez-Bautista VM, Yamazaki-Nakashimada MA (September 2012). "Fever is not always present in Kawasaki disease". Rheumatology International. 32 (9): 2953–54. doi:10.1007/s00296-011-2123-4. PMID21881982. S2CID1471650.
12Mori M, Imagawa T, Yasui K, Kanaya A, Yokota S (August 2000). "Predictors of coronary artery lesions after intravenous gamma-globulin treatment in Kawasaki disease". The Journal of Pediatrics. 137 (2): 177–80. doi:10.1067/mpd.2000.107890. PMID10931408.
↑Svobodová D, Slaný J, Pískovský T (2008). "[Kawasaki disease and its ocular manifestations]" [Kawasaki disease and its ocular manifestations]. Casopis Lekaru Ceskych (in Czech). 147 (3): 162–4. PMID18401983.
↑Bachmeyer C, Turc Y, Curan D, Duval-Arnould M (January 2000). "Anterior uveitis as the initial sign of adult Kawasaki syndrome (mucocutaneous lymph node syndrome)". American Journal of Ophthalmology. 129 (1): 101–2. doi:10.1016/S0002-9394(99)00285-8. PMID10653425.
↑Smith LB, Newburger JW, Burns JC (February 1989). "Kawasaki syndrome and the eye". The Pediatric Infectious Disease Journal. 8 (2): 116–8. PMID2468129.
12Kubota M, Usami I, Yamakawa M, Tomita Y, Haruta T (June 2008). "Kawasaki disease with lymphadenopathy and fever as sole initial manifestations". Journal of Paediatrics and Child Health. 44 (6): 359–62. doi:10.1111/j.1440-1754.2008.01310.x. PMID18476929. S2CID32280647.
↑González Pascual E, Villanueva Lamas J, Ros Viladoms J, Pons Odena M, Ruiz García-Diego S (January 1999). "Enfermedad de Kawasaki: presentación de cincuenta casos"[Kawasaki disease: A report of 50 cases](PDF). Anales Espanoles de Pediatria (in Spanish). 50 (1): 39–43. PMID10083641.
↑Kwan YW, Leung CW (December 2005). "Pustulo-vesicular skin eruption in a child with probable Kawasaki disease". European Journal of Pediatrics. 164 (12): 770–1. doi:10.1007/s00431-005-1715-y. PMID16010565. S2CID22194695.
123Fujiwara H, Fujiwara T, Kao TC, Ohshio G, Hamashima Y (June 1986). "Pathology of Kawasaki disease in the healed stage. Relationships between typical and atypical cases of Kawasaki disease". Pathology International. 36 (6): 857–67. doi:10.1111/j.1440-1827.1986.tb03119.x. PMID3766134. S2CID12989507.
12Hirose O, Misawa H, Kijima Y, Yamada O, Arakaki Y, Kajino Y, etal. (March 1981). "[Two-dimensional echocardiography of coronary artery in Kawasaki disease (MCLS): detection, changes in acute phase, and follow-up observation of the aneurysm (author's transl)]". Journal of Cardiography (in Japanese). 11 (1): 89–104. PMID7264399.
↑Burns JC, Wiggins JW, Toews WH, Newburger JW, Leung DY, Wilson H, Glodé MP (November 1986). "Clinical spectrum of Kawasaki disease in infants younger than 6 months of age". The Journal of Pediatrics. 109 (5): 759–63. doi:10.1016/S0022-3476(86)80689-8. PMID3772656.
↑Boven K, De Graeff-Meeder ER, Spliet W, Kuis W (August 1992). "Atypical Kawasaki disease: an often missed diagnosis". European Journal of Pediatrics. 151 (8): 577–80. doi:10.1007/BF01957725. PMID1505575. S2CID6125622.
↑Suzuki A, Kamiya T, Kuwahara N, Ono Y, Kohata T, Takahashi O, etal. (1986). "Coronary arterial lesions of Kawasaki disease: cardiac catheterization findings of 1100 cases". Pediatric Cardiology. 7 (1): 3–9. doi:10.1007/BF02315475. PMID3774580. S2CID20301847.
↑Terai M, Shulman ST (December 1997). "Prevalence of coronary artery abnormalities in Kawasaki disease is highly dependent on gamma globulin dose but independent of salicylate dose". The Journal of Pediatrics. 131 (6): 888–93. doi:10.1016/S0022-3476(97)70038-6. PMID9427895.
↑Kobayashi T, Inoue Y, Morikawa A (February 2008). "[Risk stratification and prediction of resistance to intravenous immunoglobulin in Kawasaki disease]". Nihon Rinsho. Japanese Journal of Clinical Medicine (in Japanese). 66 (2): 332–7. PMID18260333.
12Koren G, Lavi S, Rose V, Rowe R (March 1986). "Kawasaki disease: review of risk factors for coronary aneurysms". The Journal of Pediatrics. 108 (3): 388–92. doi:10.1016/S0022-3476(86)80878-2. PMID3950818.
123Kato H, Ichinose E, Kawasaki T (June 1986). "Myocardial infarction in Kawasaki disease: clinical analyses in 195 cases". The Journal of Pediatrics. 108 (6): 923–7. doi:10.1016/S0022-3476(86)80928-3. PMID3712157.
↑Suzuki A, Kamiya T, Tsuchiya K, Sato I, Arakaki Y, Kohata T, Ono Y (February 1988). "Tricuspid and mitral regurgitation detected by color flow Doppler in the acute phase of Kawasaki disease". The American Journal of Cardiology. 61 (4): 386–90. doi:10.1016/0002-9149(88)90950-2. PMID3341217.
↑Akagi T, Kato H, Inoue O, Sato N, Imamura K (August 1990). "Valvular heart disease in Kawasaki syndrome: incidence and natural history". American Heart Journal. 120 (2): 366–72. doi:10.1016/0002-8703(90)90081-8. PMID2382613.
↑Fukunaga S, Egashira A, Arinaga K, Akasu I, Kai E, Higashi T, etal. (March 1996). "Aortic valve replacement for aortic regurgitation due to Kawasaki disease. Report of two cases". The Journal of Heart Valve Disease. 5 (2): 231–4. PMID8665019.
↑Fuyama Y, Hamada R, Uehara R, Yano I, Fujiwara M, Matoba M, etal. (June 1996). "Long-term follow up of abdominal aortic aneurysm complicating Kawasaki disease: comparison of the effectiveness of different imaging methods". Acta Paediatrica Japonica. 38 (3): 252–5. doi:10.1111/j.1442-200X.1996.tb03480.x. PMID8741316. S2CID30968335.
↑Miyake T, Yokoyama T, Shinohara T, Seto S, Oiki M (August 1995). "Transient dilatation of the abdominal aorta in an infant with Kawasaki disease associated with thrombocytopenia". Acta Paediatrica Japonica. 37 (4): 521–5. doi:10.1111/j.1442-200X.1995.tb03368.x. PMID7572158. S2CID30747089.
↑Ooyanagi R, Fuse S, Tomita H, Takamuro M, Horita N, Mori M, Tsutsumi H (August 2004). "Pulse wave velocity and ankle brachial index in patients with Kawasaki disease". Pediatrics International. 46 (4): 398–402. doi:10.1111/j.1442-200x.2004.01929.x. PMID15310302. S2CID21586626.
↑Yaniv L, Jaffe M, Shaoul R (September 2005). "The surgical manifestations of the intestinal tract in Kawasaki disease". Journal of Pediatric Surgery. 40 (9): e1–4. doi:10.1016/j.jpedsurg.2005.05.063. PMID16150324.
↑Beiler HA, Schmidt KG, von Herbay A, Löffler W, Daum R (April 2001). "Ischemic small bowel strictures in a case of incomplete Kawasaki disease". Journal of Pediatric Surgery. 36 (4): 648–50. doi:10.1053/jpsu.2001.22311. PMID11283899.
↑Zulian F, Falcini F, Zancan L, Martini G, Secchieri S, Luzzatto C, Zacchello F (June 2003). "Acute surgical abdomen as presenting manifestation of Kawasaki disease". The Journal of Pediatrics. 142 (6): 731–5. doi:10.1067/mpd.2003.232. PMID12838207.
↑Ohno S, Miyajima T, Higuchi M, Yoshida A, Matsuda H, Saheki Y, etal. (June 1982). "Ocular manifestations of Kawasaki's disease (mucocutaneous lymph node syndrome)". American Journal of Ophthalmology. 93 (6): 713–7. doi:10.1016/0002-9394(82)90465-2. PMID7201245.
↑Burke MJ, Rennebohm RM (1981). "Eye involvement in Kawasaki disease". Journal of Pediatric Ophthalmology and Strabismus. 18 (5): 7–11. doi:10.3928/0191-3913-19810901-04. PMID7299613.
↑Anand S, Yang YC (2004). "Optic disc changes in Kawasaki disease". Journal of Pediatric Ophthalmology and Strabismus. 41 (3): 177–9. doi:10.3928/0191-3913-20040501-12. PMID15206604.
↑Farvardin M, Kashef S, Aleyasin S, Nabavizadeh SH, Sajjadi M, Safari M (2007). "Sudden unilateral blindness in a girl with Kawasaki disease". Journal of Pediatric Ophthalmology and Strabismus. 44 (5): 303–04. doi:10.3928/01913913-20070901-06. PMID17913174.
↑Tomita S, Chung K, Mas M, Gidding S, Shulman ST (January 1992). "Peripheral gangrene associated with Kawasaki disease". Clinical Infectious Diseases. 14 (1): 121–6. doi:10.1093/clinids/14.1.121. PMID1571415.
↑Takagi K, Umezawa T, Saji T, Morooka K, Matsuo N (September 1990). "[Meningoencephalitis in Kawasaki disease]". No to Hattatsu = Brain and Development (in Japanese). 22 (5): 429–35. PMID2223179.
↑Aoki N (March 1988). "Subdural effusion in the acute stage of Kawasaki disease (Mucocutaneous lymph node syndrome)". Surgical Neurology. 29 (3): 216–7. doi:10.1016/0090-3019(88)90009-2. PMID3344468.
↑Bailie NM, Hensey OJ, Ryan S, Allcut D, King MD (2001). "Bilateral subdural collections--an unusual feature of possible Kawasaki disease". European Journal of Paediatric Neurology. 5 (2): 79–81. doi:10.1053/ejpn.2001.0469. PMID11589317.
↑Silva CH, Roscoe IC, Fernandes KP, Novaes RM, Lázari CS (2002). "[Sensorineural hearing loss associated to Kawasaki Disease]". Jornal de Pediatria (in English and Portuguese). 78 (1): 71–74. doi:10.2223/JPED.669 (inactive 14 August 2025). PMID14647816.{{cite journal}}: CS1 maint: DOI inactive as of August 2025 (link)
↑Shulman, Stanford T.; Taubert, Kathryn A. (June 1999). "Kawasaki Disease". American Family Physician. 59 (11): 3093–102, 3107–08. PMID10392592. Archived from the original on 17 May 2008.
12345Guillevin L, Pagnoux C (March 2008). "[Classification of systemic vasculitides]". La Revue du Praticien (in French). 58 (5): 480–86. PMID18524103.
↑Rigante D (2006). "Clinical overview of vasculitic syndromes in the pediatric age". European Review for Medical and Pharmacological Sciences. 10 (6): 337–45. PMID17274537. S2CID15223179.
↑Hamada H, Suzuki H, Onouchi Y, Ebata R, Terai M, Fuse S, Okajima Y, Kurotobi S, Hirai K, Soga T, Ishiguchi Y, Okuma Y, Takada N, Yanai M, Sato J, Nakayashiro M, Ayusawa M, Yamamoto E, Nomura Y, Hashimura Y, Ouchi K, Masuda H, Takatsuki S, Hirono K, Ariga T, Higaki T, Otsuki A, Terauchi M, Aoyagi R, Sato T, Fujii Y, Fujiwara T, Hanaoka H, Hata A, KAICA trial Investigators (March 2019). "Efficacy of primary treatment with immunoglobulin plus ciclosporin for prevention of coronary artery abnormalities in patients with Kawasaki disease predicted to be at increased risk of non-response to intravenous immunoglobulin (KAICA): a randomised controlled, open-label, blinded-endpoints, phase 3 trial". The Lancet. 393 (10176): 1128–1137. doi:10.1016/S0140-6736(18)32003-8. PMID30853151. S2CID72335365.
↑Sundel RP, Baker AL, Fulton DR, Newburger JW (June 2003). "Corticosteroids in the initial treatment of Kawasaki disease: report of a randomized trial". The Journal of Pediatrics. 142 (6): 611–16. doi:10.1067/mpd.2003.191. PMID12838187.
↑Beiser AS, Takahashi M, Baker AL, Sundel RP, Newburger JW (May 1998). "A predictive instrument for coronary artery aneurysms in Kawasaki disease. US Multicenter Kawasaki Disease Study Group". The American Journal of Cardiology. 81 (9): 1116–20. doi:10.1016/S0002-9149(98)00116-7. PMID9605052.
↑Fujiwara T, Fujiwara H, Hamashima Y (1987). "Size of coronary aneurysm as a determinant factor of the prognosis in Kawasaki disease: clinicopathologic study of coronary aneurysms". Progress in Clinical and Biological Research. 250: 519–20. PMID3423060.
↑Nakano H, Ueda K, Saito A, Nojima K (November 1985). "Repeated quantitative angiograms in coronary arterial aneurysm in Kawasaki disease". The American Journal of Cardiology. 56 (13): 846–51. doi:10.1016/0002-9149(85)90767-2. PMID4061324.
↑Tatara K, Kusakawa S (November 1987). "Long-term prognosis of giant coronary aneurysm in Kawasaki disease: an angiographic study". The Journal of Pediatrics. 111 (5): 705–10. doi:10.1016/S0022-3476(87)80246-9. PMID3668739.
↑Ishii M, Ueno T, Akagi T, Baba K, Harada K, Hamaoka K, etal. (October 2001). "Guidelines for catheter intervention in coronary artery lesion in Kawasaki disease". Pediatrics International. 43 (5): 558–62. doi:10.1046/j.1442-200X.2001.01464.x. PMID11737728. S2CID39330448.
↑Akagi T, Ogawa S, Ino T, Iwasa M, Echigo S, Kishida K, etal. (August 2000). "Catheter interventional treatment in Kawasaki disease: A report from the Japanese Pediatric Interventional Cardiology Investigation group". The Journal of Pediatrics. 137 (2): 181–86. doi:10.1067/mpd.2000.107164. PMID10931409.
↑Holman, R. C. (2003). "Kawasaki Syndrome Hospitalizations in the United States, 1997 and 2000". Pediatrics. 112 (3): 495–501. doi:10.1542/peds.112.3.495. PMID12949272.
↑Yamamoto T, Oya T, Watanabe A (1968). "Clinical features of Kawasaki disease". Shonika Rinsho (in Japanese). 21: 291–97.
↑Kawasaki T, Kosaki F, Okawa S, Shigematsu I, Yanagawa H (September 1974). "A new infantile acute febrile mucocutaneous lymph node syndrome (MLNS) prevailing in Japan". Pediatrics. 54 (3): 271–76. doi:10.1542/peds.54.3.271. PMID4153258. S2CID13221240.
↑Melish ME, Hicks RM, Larson EJ (June 1976). "Mucocutaneous lymph node syndrome in the United States". American Journal of Diseases of Children. 130 (6): 599–607. doi:10.1001/archpedi.1976.02120070025006. PMID7134.
↑Taubert KA, Rowley AH, Shulman ST (1995). "A 10 year (1984–1993) United States hospital survey of Kawasaki disease". In Kato H (ed.). Kawasaki disease: Proceedings of the 5th International Kawasaki Disease Symposium, Fukuoka, Japan, 22–25 May 1995. Vol.1093. Elsevier. pp.34–38. ISBN0-444-82200-3. ISSN0531-5131.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.





{kind=link}
{kind=link}