Early life and education
Born in Chicago, Illinois, to a Jewish [10] family, Beutler lived in Southern California between the ages of 2 and 18 (1959 to 1977). For most of this time, he lived in city of Arcadia, a northeastern suburb of Los Angeles in the San Gabriel Valley. During these years, he spent much time hiking in the San Gabriel Mountains, and in regional national parks (Sequoia, Yosemite, Joshua Tree, and Grand Canyon), and was particularly fascinated by living things. [11] These experiences impelled an intense interest in biological science. His introduction to experimental biology, acquired between the ages of 14 and 18, included work in the laboratory of his father, Ernest Beutler, then at the City of Hope Medical Center in Duarte, CA. There he learned to assay enzymes of red blood cells and became familiar with methods for protein isolation. He published his studies of an electrophoretic variant of glutathione peroxidase, [12] as well as the inherent catalytic activity of inorganic selenite, [13] at the age of 17.
Beutler also worked in the City of Hope laboratory of Susumu Ohno, a geneticist known for his studies of evolution, genome structure, and sex differentiation in mammals. Ohno hypothesized that the major histocompatibility complex proteins served as anchorage sites for organogenesis-directing proteins. [14] He further suggested that H-Y antigen, a minor histocompatibility protein encoded by a gene on the Y chromosome and absent in female mammals, was responsible for directing organogenesis of the indifferent gonad to form a testis. In studying H-Y antigen, [15] Beutler became conversant with immunology and mouse genetics during the 1970s. While a college student at the University of California at San Diego, Beutler worked in the laboratory of Dan Lindsley, a Drosophila geneticist interested in spermatogenesis and spermiogenesis in the fruit fly. There, he learned to map phenotypes to chromosomal regions using visible phenotypic markers. [11] He also worked in the laboratory of Abraham Braude, an expert in the biology of LPS.
Beutler received his secondary school education at Polytechnic School in Pasadena, California. A precocious student, he graduated from high school at the age of 16, enrolled in college at the University of California, San Diego, and graduated with a BA degree at the age of 18 in 1976. He then enrolled in medical school at the University of Chicago in 1977 and received his M.D. degree in 1981 at the age of 23. [16] From 1981 to 1983 Beutler continued his medical training at the University of Texas Southwestern Medical Center in Dallas, Texas, as an intern in the Department of Internal Medicine, and as a resident in the Department of Neurology. However, he found clinical medicine less interesting than laboratory science, and decided to return to the laboratory.
Scientific contributions
Beutler’s focus on innate immunity began when he was a postdoctoral associate and later an assistant professor in the lab of Anthony Cerami at Rockefeller University (1983-1986). Drawing upon skills he had acquired earlier, he isolated mouse “cachectin” from the conditioned medium of LPS-activated mouse macrophages. [17] Cachectin was hypothesized by Cerami to be a mediator of wasting in chronic disease. Its biological activity, the suppression of lipoprotein lipase synthesis in adipocytes, was thought to contribute to wasting, since lipoprotein lipase cleaves fatty acids from circulating triglycerides, allowing their uptake and re-esterification within fat cells. [18] By sequential fractionation of LPS-activated macrophage medium, measuring cachectin activity at each step, Beutler purified cachectin to homogeneity. [19] Determining its N-terminal sequence, he recognized it as mouse tumor necrosis factor (TNF), and showed that it had strong TNF activity; moreover that human TNF, isolated by a very different assay, had strong cachectin activity. [18]
Human TNF, isolated contemporaneously by other workers, [20] had to that time been defined only by its ability to kill cancer cells. The discovery of a separate role for TNF as a catabolic switch was of considerable interest. Of still greater importance, Beutler demonstrated that TNF acted as a key mediator of endotoxin-induced shock. [21] This he accomplished by raising an antibody against mouse TNF, which he used to neutralize TNF in living mice challenged with lipopolysaccharide (LPS). [21] The often-lethal systemic inflammatory response to LPS was significantly mitigated by passive immunization against TNF. The discovery that TNF caused an acute systemic inflammatory disease (LPS-induced shock) presaged its causative role in numerous chronic inflammatory diseases. With J.-M. Dayer, Beutler demonstrated that purified TNF could cause inflammation-associated responses in cultured human synoviocytes: secretion of collagenase and prostaglandin E2. [22] This was an early hint that TNF might be causally important in rheumatoid arthritis (as later shown by Feldmann, Brennan, and Maini [23] ). Beutler also demonstrated the existence of TNF receptors on most cell types, [19] and correctly inferred the presence of two types of TNF receptor distinguished by their affinities, later cloned and designated p55 and p75 TNF receptors to denote their approximate molecular weights. [24] [25] [26] [27] [28] Before a sensitive immunoassay for TNF was feasible, Beutler used these receptors in a binding competition assay using radio-iodinated TNF as a tracer, which allowed him to precisely measure TNF in biological fluids. [29]
Invention of TNF inhibitors
Beutler was recruited to a faculty position at UT Southwestern Medical Center and the Howard Hughes Medical Institute in 1986. Aware that TNF blockade might have clinical applications, he (along with a graduate student, David Crawford, and a postdoctoral associate, Karsten Peppel) invented and patented recombinant molecules expressly designed to neutralize TNF in vivo (Patent No. US5447851B1). [30] Fusing the binding portion of TNF receptor proteins to the heavy chain of an immunoglobulin molecule to force receptor dimerization, [30] they produced chimeric reagents with surprisingly high affinity and specificity for both TNF and a closely related cytokine called lymphotoxin, low antigenicity, and excellent stability in vivo. The human p75 receptor chimeric protein was later used extensively as the drug Etanercept in the treatment of rheumatoid arthritis, Crohn's disease, psoriasis, and other forms of inflammation. Marketed by Amgen, Etanercept achieved more than $74B in sales. [31]
Discovery of the LPS receptor, and the role of TLRs in innate immune sensing
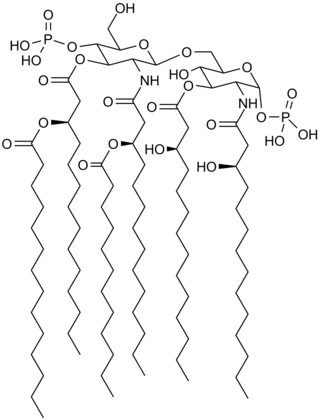
From the mid-1980s onward Beutler was interested in the mechanism by which LPS activates mammalian immune cells (chiefly macrophages, [18] [21] but dendritic cells and B cells as well), sometimes leading to uncontrollable Gram negative septic shock, [32] [33] [34] but also promoting the well-known adjuvant effect of LPS, [35] and B cell mitogenesis [36] [37] and antibody production. A single, highly specific LPS receptor was presumed to exist as early as the 1960s, based on the fact that allelic mutations in two separate strains of mice, affecting a discrete genetic locus on chromosome 4 termed Lps, abolished LPS sensing. [36] [38] Although this receptor had been widely pursued, it remained elusive. Beutler reasoned that in finding the LPS receptor, insight might be gained into the first molecular events that transpire upon an encounter between the host and microbial invaders. [39]
Utilizing positional cloning in an effort that began in 1993 and lasted five years, Beutler, together with several postdoctoral associates including Alexander Poltorak, measured TNF production as a qualitative phenotypic endpoint of the LPS response. Analyzing more than 2,000 meioses, they confined the LPS receptor-encoding gene to a region of the genome encompassing approximately 5.8 million base pairs of DNA. [2] Sequencing most of the interval, they identified a gene within which each of two LPS-refractory strains of mice (C3H/HeJ and C57BL/10ScCr) had deleterious mutations. The gene, Tlr4, encoded a cell surface protein with cytoplasmic domain homology to the interleukin-1 receptor, and several other homologous genes that were scattered across the mouse genome. Beutler and his team thus proved that one of the mammalian Toll-like receptors, TLR4, acts as the membrane-spanning component of the mammalian LPS receptor complex. [2] [40] [41] They also showed that while mouse TLR4 is activated by a tetra-acylated LPS-like molecule (lipid IVa), human TLR4 is not, recapitulating the species specificity for LPS partial structures. [41] It was deduced that direct contact between TLR4 and LPS is a prerequisite for cell activation. [41] Later, an extracellular component of the LPS receptor complex, MD-2 (also known as lymphocyte antigen 96), was identified by R. Shimazu and colleagues. [42] The structure of the complex, with and without LPS bound, was solved by Jie-Oh Lee and colleagues in 2009. [43]
Jules Hoffmann and colleagues had earlier shown that the Drosophila Toll protein, originally known for its role in embryogenesis, was essential for the antimicrobial peptide response to fungal infection. [44] However, no molecule derived from fungi actually became bound to Toll; rather, a proteolytic cascade led to the activation of an endogenous ligand, the protein Spätzle. This activated NF-kB within cells of the fat body, leading to antimicrobial peptide secretion.
Aware of this work, Charles Janeway and Ruslan Medzhitov overexpressed a modified version of human TLR4 (which they called ‘h-Toll’) and found it capable of activating the transcription factor NF-κB in mammalian cells. [45] They speculated that TLR4 was a “pattern recognition receptor.” However, they provided no evidence that TLR4 recognized any molecule of microbial origin. If a ligand did exist, it might have been endogenous (as in the fruit fly, where Toll recognizes the endogenous protein Spätzle, or as in the case of the IL-1 receptor, which recognizes the endogenous cytokine IL-1). Indeed, numerous cell surface receptors, including the TGFβ receptor, B cell receptor, and T cell receptor activate NF-κB. In short, it was not clear what TLR4 recognized, nor what its function was. Separate publications, also based on transfection/overexpression studies, held that TLR2 rather than TLR4 was the LPS receptor. [46] [47]
The genetic evidence of Beutler and coworkers correctly identified TLR4 as the specific and non-redundant cell surface receptor for LPS, fully required for virtually all LPS activities. This suggested that other TLRs (of which ten are now known to exist in humans) might also act as sensors of infection in mammals, [48] each detecting other signature molecules made by microbes whether or not they were pathogens in the classical sense of the term. The other TLRs, like TLR4, do indeed initiate innate immune responses. By promoting inflammatory signaling, TLRs can also mediate pathologic effects including fever, systemic inflammation, and shock. Sterile inflammatory and autoimmune diseases such as systemic lupus erythematosus also elicit TLR signaling, and disruption of signaling from the nucleic acid sensing TLRs can favorably modify the disease phenotype. [49] [50] [51] [52] [53] [54] [55] [56]
Random Germline Mutagenesis/Forward Genetics in the mouse
After completing the positional cloning of the Lps locus in 1998, Beutler continued to apply a forward genetic approach to the analysis of immunity in mammals. In this process, germline mutations that alter immune function are created in mice through a random process using the alkylating agent ENU, detected by their phenotypic effects, and then isolated by positional cloning. [57] This work disclosed numerous essential signaling molecules required for the innate immune response, [58] [59] [60] [61] [62] [63] [64] and helped to delineate the biochemistry of innate immunity. Among the genes detected was Ticam1, implicated by an ENU-induced phenotype called Lps2. [58] The encoded protein TICAM1, also known as TRIF, was a new adaptor molecule, binding to the cytoplasmic domains of both TLR3 and TLR4, and needed for signaling by each.
Another phenotype, called 3d to connote a “triple defect” in TLR signaling, affected a gene of unknown function called Unc93b1. [60] TLRs 3, 7, and 9 (nucleic acid sensing TLRs) failed to signal in homozygotes for the mutation. These TLRs were found to be endosomal, and physically interact with the UNC93B1 protein which transports them to the endosomal compartment. [65] Humans with mutations in UNC93B1, the human ortholog of the same gene, were subsequently found to be susceptible to recurrent Herpes simplex virus (HSV) encephalitis, in which reactivation of latent virus occurs repeatedly in the trigeminal ganglion at the base of the midbrain, leading to cortical neuron death. [66]
Yet another protein needed to make the endosomal environment suitable for TLR signaling was SLC15A4, identified based on the phenotype feeble. [67] feeble was identified in a screen in which immunostimulatory DNA was administered to mice intravenously with measurement of the systemic type I interferon response. Failure of this response, which is dependent on TLR9 signaling from plasmacytoid dendritic cells (pDC) was observed in homozygous mutants, and subsequently, failure of TLR7 (but not TLR3) signaling was observed as well. Because the feeble mutation suppressed SLE in mice, [52] the SLC15A4 protein has become a target of interest for drug development. [68]
In all, Beutler and colleagues detected 77 mutations in 36 genes in which ENU-induced mutations created defects of TLR signaling, detected due to faulty TNF and/or interferon responses. These genes encoded all TLRs kept under surveillance in screening, all of the four adapter proteins that signal from TLRs, kinases and other signaling proteins downstream, chaperones needed to escort TLRs to their destinations, proteins that promote the availability of TLR ligands, proteins involved in vesicle transport, and proteins involved in transcriptional responses to TLR signaling, or the post-translational processing of TNF and/or type I interferons (the proteins assayed in screening).
Beutler and colleagues also used ENU mutagenesis to study the global response to a defined infectious agent. They measured susceptibility to mouse cytomegalovirus (MCMV) and identified numerous genes that make a life-or-death difference during infection, terming this set of genes the MCMV "resistome". [69] [70] These genes were grouped into "sensing," "signaling," "effector," "homeostatic," and "developmental" categories, some of which were wholly unexpected. In the homeostatic category, for example, Kir6.1 ATP-sensitive potassium channels in the smooth muscle of the coronary arteries serve an essential role in the maintenance of blood flow during MCMV infection, and mutations that damage these channels cause sudden death during infection. [71]
Other genetic screens in the Beutler laboratory were used to identify genes that mediate homeostatic adaptations of the intestinal epithelium following a cytotoxic insult; [72] [73] [74] [75] [76] [77] [78] prevent allergic responses, [79] diabetes, [80] [81] or obesity; [82] [83] [84] support normal hematopoiesis; [85] [86] [87] [88] [89] [90] [91] [92] [93] [94] and enable humoral and cellular immunity. [95] [96] [97] [98] Some of these (beginning ~2015) were identified by a new process called automated meiotic mapping, which enabled greatly accelerated mutation identification compared to traditional genetic mapping (see below). In the course of their work, Beutler and his colleagues also discovered genes required for biological processes such as normal iron absorption, [99] hearing, [100] pigmentation, [101] [102] metabolism, [82] [84] [103] [104] [105] and embryonic development. [106] Many human diseases were ultimately linked to variants in the corresponding human genes after initial identification in the mouse by the Beutler laboratory, [66] [107] [108] [109] or by the laboratories of collaborating investigators. [110] [111] [112]
Invention of Automated Meiotic Mapping
Prior to 2013, despite the development of methods for massively parallel sequencing and their application in finding induced germline mutations, [113] [114] [115] positional cloning remained a slow process, limited by the need to genetically map mutations to chromosomal intervals to ascertain which induced mutation (among the average of approximately 60 changes in coding and splicing function induced per pedigree) was responsible for an observed phenotype. This required expansion of a mutant stock, outcrossing to a mapping strain, backcrossing, and genotypic and phenotypic analysis of F2 offspring. Moreover, when phenotypic screening was performed prior to positional cloning, only large effect size mutations (producing essentially qualitative phenotypes) were recoverable.
Beutler invented a means of instantly identifying ENU-induced mutations that cause phenotypes. [116] The process, called automated meiotic mapping (AMM), eliminates the need to breed mutant mice to a mapping strain as required in classical genetic mapping and flags causative mutations as soon as phenotypic assay data are collected. In a laboratory setting, it accelerates positional cloning approximately 200-fold, and permits ongoing measurement of genome saturation as mutagenesis progresses. [117] Not only qualitative phenotypes, but subtle quantitative phenotypes, are detectable and mapped to individual mutations; hence the sensitivity of forward genetics is dramatically increased. AMM depends on statistical computation to detect associations between mutations in either the homozygous or heterozygous state and deviant phenotypes. [116] In addition, machine learning software, trained on the outcome of many thousands of experiments in which putative causative mutations were re-created and re-assayed for phenotype, is used to assess data quality. [118] As of 2022, more than 260,000 ENU-induced non-synonymous coding or splice site mutations had been assayed for phenotypic effects, and more than 5,800 mutations in approximately 2,500 genes had been declared causative of phenotype(s). For certain screens, such as flow cytometry performed on the blood of germline mutant mice, more than 55% saturation of the genome has been achieved (i.e., more than 55% of all genes in which mutations will create flow cytometric aberrations in the peripheral blood have been detected, most of them based on assessment of multiple alleles, as of July 2021). [118]
AMM led to the discovery of many new immunodeficiency disorders, [88] [89] [90] [91] [92] [93] [94] [85] [98] and disorders of bone morphology or mineral density, [111] [112] vision, [119] and metabolism. [82] [84] [104] [105] Of note, AMM was used in the identification of a chemosensor that mediates innate fear behavior in mice and an autism gene found first in mice and then shown to cause autism in humans. [110] [120] AMM has also permitted high speed searches for mutations that suppress or augment disease phenotypes; for example, the development of autoimmune (Type 1) diabetes in mice of the NOD strain. [80] [81] It offers a rational way to investigate the pathogenesis of complex disease phenotypes in general, in which many loci invariably contribute to susceptibility or resistance to disease, and disease occurs in those individuals with an unfavorable imbalance between these opposing influences.
Developing drugs that activate TLRs
Beutler has collaborated with Dale L. Boger and his research group to identify synthetic small molecule agonists of mammalian TLRs, which may be used in combination with defined molecular antigens to precisely target and coordinate innate and adaptive immune responses. Neoseptins, small molecules with no relationship to the structure of LPS, were shown to bind to the TLR4-MD2 complex in such a manner that two drug molecules trigger a conformational change similar to that elicited by an authentic LPS molecule. Diprovocims, which bear no structural similarity to bacterial lipopeptides, activate the TLR1-TLR2 heterodimer complex that normally acts as a receptor for tri-acylated lipopeptide molecules. These studies demonstrated that TLR2 and TLR4 can indeed respond to molecules other than classical microbial ligands, and set a new standard for verifying such interactions, in that X-ray crystallography was used to demonstrate the binding of neoseptins and diprovocims to their respective TLR targets at atomic level resolution. Beutler and colleagues also showed, again using X-ray crystallography combined with biological assays, that endogenous sulfatides are capable of binding to the TLR4-MD2 complex, causing its activation. [121] [122] [123] [124] [125] [126] [127]
Family
Bruce Beutler was the third son of Ernest Beutler (1928-2008) and Brondelle May Beutler (née Fleisher; 1928-2019). His siblings included two older brothers (Steven [b. 1952] and Earl [b. 1954]), and a younger sister, Deborah [b. 1962]). [133]
Ernest Beutler was a hematologist and medical geneticist famed for his studies of G-6-PD deficiency, [134] other hemolytic anemias, [135] [136] iron metabolism, [137] glycolipid storage diseases, [138] and leukemias, [139] [140] as well as his discovery of X chromosome inactivation. [141] He was a Professor and department chairman at The Scripps Research Institute contemporaneously with Bruce. The two collaborated productively on several topics prior to Ernest Beutler’s death in 2008. [12] [13] [142] [143] [144] [145]
Both of Ernest Beutler’s parents were physicians. [146] Bruce Beutler’s paternal grandmother, Kathe Beutler (née Italiener, daughter of Anna Rothstein, 1896-1999), [147] was a pediatrician, trained at the Charité hospital in Berlin, earning her medical diploma in 1923. Käthe Italiener married Alfred Beutler in 1925. Also a physician, Alfred Beutler was a cousin to the spectral physicist, Hans G. Beutler (1896-1942), who worked at the Kaiser Wilhelm Institute and the University of Berlin before emigrating to the USA in 1936. He continued his work at the University of Chicago until his death. [148]
Bruce Beutler married Barbara Beutler (née Lanzl) in 1980 and divorced in 1988. Three sons were born to the couple. [149] [150] [11]