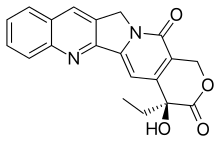
Epirubicin is an anthracycline drug used for chemotherapy. It can be used in combination with other medications to treat breast cancer in patients who have had surgery to remove the tumor. It is marketed by Pfizer under the trade name Ellence in the US and Pharmorubicin or Epirubicin Ebewe elsewhere.

Irinotecan, sold under the brand name Camptosar among others, is an anti-cancer medication used to treat colon cancer and small cell lung cancer. For colon cancer it is used either alone or with fluorouracil. For small cell lung cancer it is used with cisplatin. It is given intravenously.

Novobiocin, also known as albamycin, is an aminocoumarin antibiotic that is produced by the actinomycete Streptomyces niveus, which has recently been identified as a subjective synonym for S. spheroides a member of the class Actinomycetia. Other aminocoumarin antibiotics include clorobiocin and coumermycin A1. Novobiocin was first reported in the mid-1950s.

Topotecan, sold under the brand name Hycamtin among others, is a chemotherapeutic agent medication that is a topoisomerase inhibitor. It is a synthetic, water-soluble analog of the natural chemical compound camptothecin. It is used in the form of its hydrochloride salt to treat ovarian cancer, lung cancer and other cancer types.
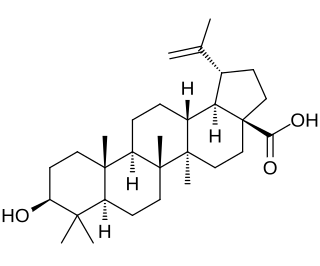
Betulinic acid is a naturally occurring pentacyclic triterpenoid which has antiretroviral, antimalarial, and anti-inflammatory properties, as well as a more recently discovered potential as an anticancer agent, by inhibition of topoisomerase. It is found in the bark of several species of plants, principally the white birch from which it gets its name, but also the ber tree, selfheal, the tropical carnivorous plants Triphyophyllum peltatum and Ancistrocladus heyneanus, Diospyros leucomelas, a member of the persimmon family, Tetracera boiviniana, the jambul, flowering quince, rosemary, and Pulsatilla chinensis.
Topoisomerase inhibitors are chemical compounds that block the action of topoisomerases, which are broken into two broad subtypes: type I topoisomerases (TopI) and type II topoisomerases (TopII). Topoisomerase plays important roles in cellular reproduction and DNA organization, as they mediate the cleavage of single and double stranded DNA to relax supercoils, untangle catenanes, and condense chromosomes in eukaryotic cells. Topoisomerase inhibitors influence these essential cellular processes. Some topoisomerase inhibitors prevent topoisomerases from performing DNA strand breaks while others, deemed topoisomerase poisons, associate with topoisomerase-DNA complexes and prevent the re-ligation step of the topoisomerase mechanism. These topoisomerase-DNA-inhibitor complexes are cytotoxic agents, as the un-repaired single- and double stranded DNA breaks they cause can lead to apoptosis and cell death. Because of this ability to induce apoptosis, topoisomerase inhibitors have gained interest as therapeutics against infectious and cancerous cells.

Helenalin, or (-)-4-Hydroxy-4a,8-dimethyl-3,3a,4a,7a,8,9,9a-octahydroazuleno[6,5-b]furan-2,5-dione, is a toxic sesquiterpene lactone which can be found in several plants such as Arnica montana and Arnica chamissonis Helenalin is responsible for the toxicity of the Arnica spp. Although toxic, helenalin possesses some in vitro anti-inflammatory and anti-neoplastic effects. Helenalin can inhibit certain enzymes, such as 5-lipoxygenase and leukotriene C4 synthase. For this reason the compound or its derivatives may have potential medical applications.

A mitotic inhibitor, microtubule inhibitor, or tubulin inhibitor, is a drug that inhibits mitosis, or cell division, and is used in treating cancer, gout, and nail fungus. These drugs disrupt microtubules, which are structures that pull the chromosomes apart when a cell divides. Mitotic inhibitors are used in cancer treatment, because cancer cells are able to grow through continuous division that eventually spread through the body (metastasize). Thus, cancer cells are more sensitive to inhibition of mitosis than normal cells. Mitotic inhibitors are also used in cytogenetics, where they stop cell division at a stage where chromosomes can be easily examined.

DNA topoisomerase 1 is an enzyme that in humans is encoded by the TOP1 gene. It is a DNA topoisomerase, an enzyme that catalyzes the transient breaking and rejoining of a single strand of DNA.
Strictosidine synthase (EC 4.3.3.2) is an enzyme in alkaloid biosynthesis that catalyses the condensation of tryptamine with secologanin to form strictosidine in a formal Pictet–Spengler reaction:
CRLX101 is an experimental approach to cancer chemotherapy that is under investigation in human trials. It is an example of a nanomedicine.
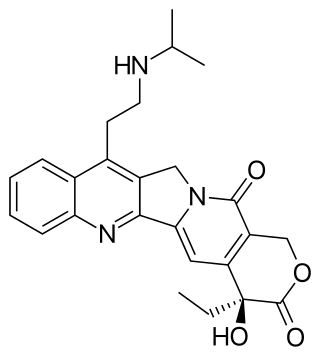
Belotecan is a drug used in chemotherapy. It is a semi-synthetic camptothecin analogue indicated for small-cell lung cancer and ovarian cancer, approved in South Korea under the trade name Camtobell, presented in 2 mg vials for injection. The drug has been marketed by Chong Kun Dang Pharmaceuticals since 2003.
The duocarmycins are members of a series of related natural products first isolated from Streptomyces bacteria in 1978. They are notable for their extreme cytotoxicity and thus represent a class of exceptionally potent antitumour antibiotics.

Strictosidine is a natural chemical compound and is classified as a glucoalkaloid and a vinca alkaloid. It is formed by the Pictet–Spengler condensation reaction of tryptamine with secologanin, catalyzed by the enzyme strictosidine synthase. Thousands of strictosidine derivatives are sometimes referred to by the broad phrase of monoterpene indole alkaloids. Strictosidine is an intermediate in the biosynthesis of numerous pharmaceutically valuable metabolites including quinine, camptothecin, ajmalicine, serpentine, vinblastine, vincristine and mitragynine.
Curacin A is a hybrid polyketide synthase (PKS)/nonribosomal peptide synthase (NRPS) derived natural product produced isolated from the cyanobacterium Lyngbya majuscula. Curacin A belongs to a family of natural products including jamaicamide, mupirocin, and pederin that have an unusual terminal alkene. Additionally, Curacin A contains a notable thiazoline ring and a unique cyclopropyl moiety, which is essential to the compound's biological activity. Curacin A has been characterized as potent antiproliferative cytotoxic compound with notable anticancer activity for several cancer lines including renal, colon, and breast cancer. Curacin A has been shown to interact with colchicine binding sites on tubulin, which inhibits microtubule polymerization, an essential process for cell division and proliferation.

Spirorenone (INN) is a steroidal antimineralocorticoid of the spirolactone group that was never marketed. Spirorenone possesses 5–8 times the antimineralocorticoid activity of spironolactone in animal studies. The initial discovery of spirorenone was deemed a great success, as no compound with greater antimineralocorticoid activity had been developed since spironolactone in 1957. Moreover, spirorenone itself has virtually no affinity for the androgen receptor while its progestogenic activity shows species differences, being somewhat greater than that of spironolactone in rabbits but absent in mice and rats. As such, it was characterized as a highly potent antimineralocorticoid with far fewer hormonal side effects relative to spironolactone.

Lipase inhibitors belong to a drug class that is used as an antiobesity agent. Their mode of action is to inhibit gastric and pancreatic lipases, enzymes that play an important role in the digestion of dietary fat. Lipase inhibitors are classified in the ATC-classification system as A08AB . Numerous compounds have been either isolated from nature, semi-synthesized, or fully synthesized and then screened for their lipase inhibitory activity but the only lipase inhibitor on the market is orlistat . Lipase inhibitors have also shown anticancer activity, by inhibiting fatty acid synthase.
Fostriecin is a type I polyketide synthase (PKS) derived natural product, originally isolated from the soil bacterium Streptomyces pulveraceus. It belongs to a class of natural products which characteristically contain a phosphate ester, an α,β-unsaturated lactam and a conjugated linear diene or triene chain produced by Streptomyces. This class includes structurally related compounds cytostatin and phoslactomycin. Fostriecin is a known potent and selective inhibitor of protein serine/threonine phosphatases, as well as DNA topoisomerase II. Due to its activity against protein phosphatases PP2A and PP4 which play a vital role in cell growth, cell division, and signal transduction, fostriecin was looked into for its antitumor activity in vivo and showed in vitro activity against leukemia, lung cancer, breast cancer, and ovarian cancer. This activity is thought to be due to PP2A's assumed role in regulating apoptosis of cells by activating cytotoxic T-lymphocytes and natural killer cells involved in tumor surveillance, along with human immunodeficiency virus-1 (HIV-1) transcription and replication.

Bisphosphonates are an important class of drugs originally commercialised in the mid to late 20th century. They are used for the treatment of osteoporosis and other bone disorders that cause bone fragility and diseases where bone resorption is excessive. Osteoporosis is common in post-menopausal women and patients in corticosteroid treatment where biphosphonates have been proven a valuable treatment and also used successfully against Paget's disease, myeloma, bone metastases and hypercalcemia. Bisphosphonates reduce breakdown of bones by inhibiting osteoclasts, they have a long history of use and today there are a few different types of bisphosphonate drugs on the market around the world.

Diflomotecan is a chemotherapeutic agent that is a topoisomerase inhibitor (Top1). It varies from other camptothecin based Top1 inhibitors like topotecan in having a 7-membered E-ring. The oxepan-2-one ring in the homocamptothecin analogues is more stable in plasma compared to the 6-membered lactone in camptothecin. Diflomotecan was the first homocamptothecin to enter clinical trials and is currently in phase I for treating patients with sensitive small cell lung cancer (SCLC).