Steroidal aromatase inhibitors are a class of drugs that are mostly used for treating breast cancer in postmenopausal women. High levels of estrogen in breast tissue increases the risk of developing breast cancer and the enzymearomatase is considered to be a good therapeutic target when treating breast cancer due to it being involved in the final step of estrogen biosynthetic pathway and also its inhibition will not affect production of other steroids. Aromatase Inhibitors are classified into two categories based on their structure, nonsteroidal and steroidal; the latter resemble the structure of androstenedione.[1] Steroidal aromatase inhibitors irreversibly inhibit the enzyme by binding covalently to the binding site of aromatase so the substrate cannot access it.[2]
Figure 1: The steroid scaffold. Base structure of steroids.
Collaboration leading to discovery
In 1944 the Worcester Foundation for Experimental Biology (WFEB) was established. The foundation was the center point of collaboration of many scientists interested in reproduction, neurophysiology and steroid biochemistry. One of the foundations work was the understanding of the mechanism of conversion of androgens to estrogens. The group worked on understanding the biosynthesis and metabolism of steroids that are produced by adrenal glands, testes and ovaries. Andre Meyer later hypothesized that the aromatization of androstenedione was enzymatic and this was proven in the 1980s with the purification of aromatase.[3] In the early 1970s the investigators agreed upon the fact that aromatization of ring A was facilitated by a cytochrome P450-mediated enzyme, proven with the blockade of aminoglutethimide (AG, known to block P450-mediated enzymes.[3]
Harry and Angela Brodie
Harry Brodie, a chemist, joined the WFEB group and started working on understanding steriochemistry of hydrogen elimination at the C-1 position during aromatization. His mechanistic studies led him to recognizing the therapeutic potential of targeting aromatase, in the early 1970s he started the development of selective aromatase inhibitors.[3] The collaboration with his wife Angela Brodie led them to report in 1973 the first series of these compounds.[4] They made systematic structure/function studies on nearly 100 steroidal aromatase inhibitors which led to the discovery of 4-hydroxyandrostenedione (4-OH-A), a potent selective aromatase inhibitor.[3]
Shift towards breast cancer treatment and clinical trials
After the Brodies demonstrated the reduction of estrogen levels in rodents and its biological efficacy with regression of rat mammary tumors, Angela Brodie went to Rome in the fall 1981 to give a presentation about her research. At the presentation was Charles Coombes a medical oncologist who expressed his interests in conducting a clinical trial with 4-hydroxy-androstenedione (4-OH-A) to treat breast cancer.[4] The collaboration of Angela Brodie, Charles Coombes a clinical oncologist, Paul Goss a clinical oncologist and Mitch Dowsett a clinical chemist and made this possible. Further clinical development was made with the help of Ciba-Geigy (now Novartis), (4-OH-A) given the new name Formestane.[3] In 1993, Formestane was introduced as Lenatron in to the market with the indicators for advanced cancer in postmenauposal women, the first selective aromatase inhibitor to do so.[5]
Due to the unfavourable characters of 4-OH-A, poor oral bio-availability and unfavourable metabolism, a group led by E. Di Salle and P. Lombardi at Farmitalia-Carlo Erba (part of Pfizer) started working on a new selective aromatase inhibitor. The group designed, synthesized and evaluated a new novel steroid, exemestane. Exemestane went through clinical trials in the 1990s and received FDA approval in 1999, marketed as Aromasin. Indication for exemestane is advanced breast cancer in postmenopausal women, where the cancer has progressed following tamoxifen therapy. Exemestane is the first oral aromatase inactivator.[5]
Steroidal aromatase inhibitors today
Clinical use of steroidal aromatase inhibitors today is more or less limited to exemestane. Use of formestane (Lentaron) is very limited and in some countries it is not used anymore. Formestane has been superseded by newer and better inhibitors with better oral availability and fewer side effects, exemestane and the newer generation of nonsteroidal aromatase inhibitors.[4]
Clinical use
Cancer
A majority of breast cancers are hormone dependent and most of them express either estrogen receptor and/or progesterone receptor.[6][7][8] That is the reason that compounds that inhibit biosynthesis of estrogen have been researched and are now the standard adjuvant therapy for breast cancer in postmenopausal women.[6][7] It has been proven that breast cancer in postmenopausal women can be treated or prevented by modulating the estrogen receptors or its ligands and as aromatase is part of the final step in estrogen conversion it is a good target for medicine. Because aromatase catalyses the final step in estrogen conversion, inhibiting it does not have any effect on synthesis of other steroids except estrogen.[7]
In postmenopausal women the production of estrogen in the ovaries has ceased. The main source of estrogen is therefore aromatization of androgens produced by the adrenal glands.[7] Estrogen production in postmenopausal women mainly occurs in peripheral adipose tissue.[6]Brain, skin, adipose tissue, normal breast tissue and breast cancer cells have aromatase but estrogen that is synthesised in breast tissue and around the cancer cells have effect on growth of the cancer. Aromatase inhibitors stops this conversion and lowers the levels of estrogen.[7]
Treating breast cancer with aromatase inhibitors is only effective in postmenopausal women because of high levels of aromatase ligands (substrate) in ovaries of premenopausal women. By inhibiting aromatase in premenopausal women the estrogen levels are reduced for a short time but it leads to activation of the hypothalamus and the pituitary axis which promote gonadotropin secretion that causes rise in estrogen levels by stimulating the ovaries.[2][7]
A study has shown that cross resistance does not always occur between nonsteroidal aromatase Inhibitors and steroidal aromatase inhibitors.[6][8] If nonsteroidal aromatase Inhibitors are not working or patients are relapsing, then the use of steroidal aromatase inhibitors could be applied for better results before patients will be forced to switch from endocrine therapy to cytotoxicchemotherapy and therefore avoiding or delaying the side effects and complications of the latter.[6]
Fertility
Aromatase inhibitors have been used to preserve fertility by stimulate ovulation in premenopausal breast cancer survivors. By inhibiting aromatase in premenopausal women the estrogen levels are reduced temporarily which leads to increased gonadotropin secretion and stimulate ovaries and that causes rise in estrogen levels.[2][7]
Examples of agents
1st and 2nd generation
Testolactone and formestane are 1st and 2nd generation aromatase inhibitors. Formestane was the first selective aromatase inhibitor that was used for breast cancer treatment but it is not in clinical use today.[3][7]
3rd generation
Exemestane is the only steroidal 3rd generation aromatase inhibitor and it has the advantage over formestane in being more potent and can be administered orally. Clinical studies have shown that 25mg/day causes 97,9% suppression of aromatase.[2]
Figure 2: Structure of testolactone
Figure 3: Structure of formestane
Figure 4: Structure of exemestane
Mechanism of action
Estrogen plays a major part in the stimulation of breast cancer cell proliferation in hormone-dependent breast cancer. High concentrations of estrogen seem to promote the development of breast cancer. Consequently, two main approaches to control and block the pathological activity of estrogens have been developed.[9] The first approach focuses on the inhibition of estrogen action by antiestrogens, which interact with the estrogen receptors. The second focuses on directly inhibiting estrogen production by inhibiting the estrogen synthetase aromatase.[10] Steroidal aromatase inhibitors are identified as Type I inhibitors which interact with the substrate-binding site of the aromatase enzyme.[11]
Aromatase is a cytochrome P450 which catalyzes three consecutive hydroxylation reactions, converting C19 androgens to aromatic C18 estrogens. After gaining electrons from NADPH-cytochrome P450 reductase, the aromatase converts androstenedione and testosterone to estrone and estradiol, respectively. The aromatization of androgen is the terminal and rate-limiting step in estrogen synthesis. Recent studies have focused on defining the active site region of the aromatase enzyme and to evaluate the most promising reaction mechanism. Three-dimensional models of the aromatase active region have also been generated, though the exact nature of the structure has not yet been fully defined.[12]
Figure 7: General reaction of the conversion of testosterone to estradiol catalyzed by aromataseFigure 8: Catalytic mechanism of aromatase
Figure 9: Structure of 17-hydroexemestane
Steroidal structure
Drugs like exemestane and other steroidal aromatase inhibitors have a steroidal structure that competes with the natural aromatase substrate androstenedione.[11] The inhibitor must share important structural features with the endogenous substrate, and as features of the androgens, allowing them to interact with the catalytic site on the enzyme protein. This renders the steroidal aromatase inhibitors inherently selective.[13] Due to its selective inhibition it will not affect the production of the other steroids in the estrogen biosynthetic pathway.[9]
Binding to the active site
The drugs bound to the catalytic site are often metabolized to intermediates which have much higher affinity for the androgen receptor. The binding of the intermediate metabolite 17-hydroexemestane for the androgen receptor is about 100 times that of the parent compound, exemestane.[11] When the metabolite attaches to the active site on the enzyme, the enzyme initiates its typical sequence of hydroxylation, but hydroxylation produces an unbreakable covalent bond between the inhibitor and the enzyme protein. Even if all unattached parts of the inhibitor are removed, the enzyme activity of the aromatase can only be restored by new enzyme synthesis.[13] The inhibitor thereby blocks the activity of the enzyme even after the drug is cleared from circulation, thus having a lasting effect in vivo. There is no need for continued presence of the drug to maintain inhibition, which in turn reduced the chance of toxic adverse effects to the patient.[10] Due to the irreversible nature of the inhibition, the steroidal AIs are often marketed as inactivators or suicide inhibitors.[9]
Structure–activity relationships (SAR)
Planarity of the A ring is very important for the affinity of the compounds to aromatase. As can be seen in table 1, where compounds 2 and 3 show substantial inhibition and also have the same stereochemical requirements. Same effect can be observed with compound 5, with inhibition of 34,6% and its 4-keto derivative, compound 7, with inhibition of 83,3%. These differences in the structure of AIs show the importance of planarity in the A ring for interaction with the active site of aromatase.[14]
Important aspect to the binding properties of the compounds is the stereochemistry in the C-5 section where the position of the hydrogen atom can be in alfa or beta positions (pointing up or down). The 5α-epimers exhibit much greater binding properties then their 5β counterparts, as can be seen with compounds 3/4 and 5/6 in table 1. These results indicate the importance of a correct angle between the A and B-ring junction for better binding to the active site of aromatase.[14]
Table 1: Inhibition of A- and D-ring modified aromatase inhibitors
Compound
Formestan
1
2
3
4
5
6
7
8
9
10
11
Structure of inhibitor
Inhibition (%) for 2μM
99.1
11.0
95.9
96.4
14.6
34.6
10.1
83.3
8.1
72.5
16.6
17.6
Changing the D-ring cyclopentanone to a six-membered δ-lactone decreased the binding ability of the compounds. Compound 9 combined the D-ring structure of testolactone and the A-ring structure of formestane but had considerably lower inhibition of aromatase than formestane.[14]
C-4 region is important for the interaction of AIs with the binding region and hydrophilic bonds such as hydroxyl or carbonyl bonds in that position can improve interaction with aromatase.[14]
Experiments using 3-deoxy steroids, yielded that 4β,19-diol had the greatest inhibition of aromatase. This is likely the cause of two polar amino acids in the active site and underlines the importance of hydrophilic groups in the steroids for better binding properties.[15]
Synthesis
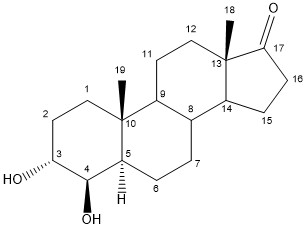
The synthesis of steroidal aromatase inhibitors is done with various methods, they all have in common that they are synthesized from a starting point that is the base structure of steroids. With various methods there a various starting point of synthesis, f.e testosterone, androstenedione and other variations of these compounds. The synthesis of Formestane from testosterone is a facile three step synthesis, as shown in figure 2. The synthesis has an overall 23% yield of Formestane. The first step is an oxidation of testosterone with Jones reagent to afford androst-4-ene-3,17-dione with a 73% yield. Step 2 is hydroxylation of androst-4-ene-3,17-dione with OsO4 followed by step 3 with alkaline dehydration of the resultant diols to give formestane.[16]
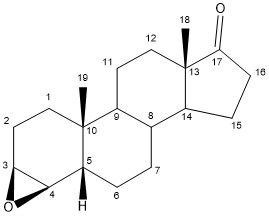
The synthesis of exemestane also consists of three steps, as shown in figure 3. First off, a Vilsmeier-Haack reagent is prepared by refluxing paraformaldehyde and dimethylamine hydrochloride in isopentanol at a temperature of 131°C while removing water from the isopentanol using a Dean-Stark separator. The internal temperature of the reagent is then cooled to 10–15°C, before adding commercially available boldenone (androsta-1,4-dien-17β-ol-3-one). The reaction mixture is then refluxed for 15 hours to give the 6-methylene derivative, 6-methyleneandrosta-1,4-dien-17β-ol-3-one. Subsequently, Jones oxidation of the derivative in acetone at −10°C gives exemestane in 79% yield after recrystallization. A mixture of 65:35 ethanol and water is used for the recrystallization process.[17]
Figure 10: Synthesis of formestaneFigure 11: Synthesis of exemestane
References
↑ Ahmad, I.; Shagufta (2015). "Recent developments in steroidal and nonsteroidal aromatase inhibitors for the chemoprevention of estrogen-dependent breast cancer". European Journal of Medicinal Chemistry. 102: 375–386. doi:10.1016/j.ejmech.2015.08.010. PMID26301554.
1 2 Lombardi, P (2002). "Exemestane, a new steroidal aromatase inhibitor of clinical relevance". Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 1587 (2–3): 326–337. doi:10.1016/S0925-4439(02)00096-0. PMID12084475.
1 2 3 4 5 Beresford, M.; Tumur, I.; Chakrabarti, J.; Barden, J.; Rao, N.; Makris, A. (2011). "A qualitative systematic review of the evidence base for non-cross-resistance between steroidal and non-steroidal aromatase inhibitors in metastatic breast cancer". Clin Oncol (R Coll Radiol). 23 (3): 209–215. doi:10.1016/j.clon.2010.11.005. PMID21134732.
1 2 3 Ahmad, Irshad (2015-09-18). "Recent developments in steroidal and nonsteroidal aromatase inhibitors for the chemoprevention of estrogen-dependent breast cancer". European Journal of Medicinal Chemistry. 102: 375–386. doi:10.1016/j.ejmech.2015.08.010. PMID26301554.
↑ Numazawa, M.; Yamada, K.; Nitta, S.; Sasaki, C.; Kidokoro, K. (2001). "Role of Hydrophilic Interaction in Binding of Hydroxylated 3-Deoxy C19 Steroids to the Active Site of Aromatase". J Med Chem. 44 (24): 4277–4283. doi:10.1021/jm010282t. PMID11708928.
↑ Li, Jie Jack (2007-01-01). Johnson, Douglas S.; Li, Jie Jack (eds.). Aromatase Inhibitors for Breast Cancer: Exemestane (Aromasin), Anastrozole (Arimidex), and Letrozole (Femara). John Wiley & Sons, Inc. pp.30–38. doi:10.1002/9780470134979.ch3. ISBN978-0-470-13497-9.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}