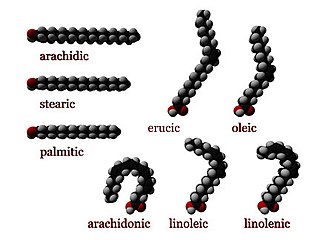
Numerous genetic disorders are caused by errors in fatty acid metabolism. These disorders may be described as fatty oxidation disorders or as a lipid storage disorders, and are any one of several inborn errors of metabolism that result from enzyme defects affecting the ability of the body to oxidize fatty acids in order to produce energy within muscles, liver, and other cell types.

Carnitine palmitoyltransferase II deficiency, sometimes shortened to CPT-II or CPT2, is an autosomal recessively inherited genetic metabolic disorder characterized by an enzymatic defect that prevents long-chain fatty acids from being transported into the mitochondria for utilization as an energy source. The disorder presents in one of three clinical forms: lethal neonatal, severe infantile hepatocardiomuscular and myopathic.

β-Hydroxybutyric acid, also known as 3-hydroxybutyric acid or BHB, is an organic compound and a beta hydroxy acid with the chemical formula CH3CH(OH)CH2CO2H; its conjugate base is β-hydroxybutyrate, also known as 3-hydroxybutyrate. β-Hydroxybutyric acid is a chiral compound with two enantiomers: D-β-hydroxybutyric acid and L-β-hydroxybutyric acid. Its oxidized and polymeric derivatives occur widely in nature. In humans, D-β-hydroxybutyric acid is one of two primary endogenous agonists of hydroxycarboxylic acid receptor 2 (HCA2), a Gi/o-coupled G protein-coupled receptor (GPCR).

Palmitoylcarnitine is an ester derivative of carnitine involved in the metabolism of fatty acids. During the tricarboxylic acid cycle (TCA), fatty acids undergo a process known as β-oxidation to produce energy in the form of ATP. β-oxidation occurs primarily within mitochondria, however the mitochondrial membrane prevents the entry of long chain fatty acids (>C10), so the conversion of fatty acids such as palmitic acid is key. Palmitic acid is brought to the cell and once inside the cytoplasm is first converted to Palmitoyl-CoA. Palmitoyl-CoA has the ability to freely pass the outer mitochondrial membrane, but the inner membrane is impermeable to the Acyl-CoA and thioester forms of various long-chain fatty acids such as palmitic acid. The palmitoyl-CoA is then enzymatically transformed into palmitoylcarnitine via the Carnitine O-palmitoyltransferase family. The palmitoylcarnitine is then actively transferred into the inner membrane of the mitochondria via the carnitine-acylcarnitine translocase. Once inside the inner mitochondrial membrane, the same Carnitine O-palmitoyltransferase family is then responsible for transforming the palmitoylcarnitine back to the palmitoyl-CoA form.

Carnitine O-palmitoyltransferase is a mitochondrial transferase enzyme involved in the metabolism of palmitoylcarnitine into palmitoyl-CoA. A related transferase is carnitine acyltransferase.
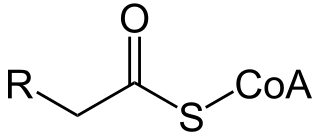
Palmitoyl-CoA is an acyl-CoA thioester. It is an "activated" form of palmitic acid and can be transported into the mitochondrial matrix by the carnitine shuttle system, and once inside can participate in beta-oxidation. Alternatively, palmitoyl-CoA is used as a substrate in the biosynthesis of sphingosine.

Carnitine palmitoyltransferase I (CPT1) also known as carnitine acyltransferase I, CPTI, CAT1, CoA:carnitine acyl transferase (CCAT), or palmitoylCoA transferase I, is a mitochondrial enzyme responsible for the formation of acyl carnitines by catalyzing the transfer of the acyl group of a long-chain fatty acyl-CoA from coenzyme A to l-carnitine. The product is often Palmitoylcarnitine, but other fatty acids may also be substrates. It is part of a family of enzymes called carnitine acyltransferases. This "preparation" allows for subsequent movement of the acyl carnitine from the cytosol into the intermembrane space of mitochondria.
The enzyme acetylsalicylate deacetylase (EC 3.1.1.55) catalyzes the reaction
The enzyme carboxylesterase (or carboxylic-ester hydrolase, EC 3.1.1.1; systematic name carboxylic-ester hydrolase) catalyzes reactions of the following form:
Palmitoyl-CoA hydrolase (EC 3.1.2.2) is an enzyme in the family of hydrolases that specifically acts on thioester bonds. It catalyzes the hydrolysis of long chain fatty acyl thioesters of acyl carrier protein or coenzyme A to form free fatty acid and the corresponding thiol:

Palmitoyl protein hydrolase/thioesterases is an enzyme (EC 3.1.2.22) that removes thioester-linked fatty acyl groups such as palmitate from modified cysteine residues in proteins or peptides during lysosomal degradation. It catalyzes the reaction
In enzymology, a retinyl-palmitate esterase (EC 3.1.1.21) is an enzyme that catalyzes the chemical reaction

Carnitine O-octanoyltransferase is a member of the transferase family, more specifically a carnitine acyltransferase, a type of enzyme which catalyzes the transfer of acyl groups from acyl-CoAs to carnitine, generating CoA and an acyl-carnitine. The systematic name of this enzyme is octanoyl-CoA:L-carnitine O-octanoyltransferase. Other names in common use include medium-chain/long-chain carnitine acyltransferase, carnitine medium-chain acyltransferase, easily solubilized mitochondrial carnitine palmitoyltransferase, and overt mitochondrial carnitine palmitoyltransferase. Specifically, CROT catalyzes the chemical reaction:

Acyl-CoA thioesterase 2, also known as ACOT2, is an enzyme which in humans is encoded by the ACOT2 gene.
Acyl-coenzyme A thioesterase 4 is an enzyme that in humans is encoded by the ACOT4 gene.

Acyl-coenzyme A thioesterase 11 also known as StAR-related lipid transfer protein 14 (STARD14) is an enzyme that in humans is encoded by the ACOT11 gene. This gene encodes a protein with acyl-CoA thioesterase activity towards medium (C12) and long-chain (C18) fatty acyl-CoA substrates which relies on its StAR-related lipid transfer domain. Expression of a similar murine protein in brown adipose tissue is induced by cold exposure and repressed by warmth. Expression of the mouse protein has been associated with obesity, with higher expression found in obesity-resistant mice compared with obesity-prone mice. Alternative splicing results in two transcript variants encoding different isoforms.
A broad classification for genetic disorders that result from an inability of the body to produce or utilize an enzyme or transport protein that is required to oxidize fatty acids. They are an inborn error of lipid metabolism, and when it affects the muscles also a metabolic myopathy.
Acyl-CoA thioesterase 6 is a protein that in humans is encoded by the ACOT6 gene. The protein, also known as C14orf42, is an enzyme with thioesterase activity.
Acyl-CoA thioesterase 1 is a protein that in humans is encoded by the ACOT1 gene.

β-Hydroxy β-methylbutyryl-coenzyme A (HMB-CoA), also known as 3-hydroxyisovaleryl-CoA, is a metabolite of L-leucine that is produced in the human body. Its immediate precursors are β-hydroxy β-methylbutyric acid (HMB) and β-methylcrotonoyl-CoA (MC-CoA). It can be metabolized into HMB, MC-CoA, and HMG-CoA in humans.
